Introduction
Myelodysplastic syndromes (MDS) encompass a heterogeneous group of disorders characterized by ineffective haematopoiesis typically resulting in cytopenias. MDS is clearly a disease of ageing patients and will be commonly encountered in primary care and geriatric practices. MDS may behave as an indolent disease or progress to bone marrow failure or acute leukaemia. Acute leukaemias are clonal disorders characterized by a proliferation of immature myeloid [acute myeloid leukaemia (AML)] or lymphoid [acute lymphocytic leukaemia (ALL)] cells. Accumulation of leukaemic cells impairs the normal haematopoietic function of the bone marrow, resulting in cytopenias with or without leukocytosis. While relatively uncommon, the incidence of acute leukaemias, particularly AML, increases with age. In addition, management issues are increasingly complex in older adults due to the aggressive nature of both the disease and the treatments required for cure. This chapter outlines key diagnosis and management issues for older adults with MDS and acute leukaemias.
Myelodysplastic Syndromes
The myelodysplastic syndromes (MDS) represent a broad group of clonal haematopoietic disorders characterized by ineffective haematopoiesis resulting in peripheral blood cytopenias. In these diseases, cells of the affected lineage are unable to undergo maturation and differentiation, resulting in cytopenias. The major clinical significance is the morbidity associated with profound cytopenias and the potential to evolve into AML.
MDS are clearly diseases of ageing patients, with a median age at diagnosis of 76 years.1 It is estimated that over 80% of the 10–15 000 patients diagnosed in 2010 will be over age 60 years.2 Unfortunately, survival for MDS has been poor with a reported three-year overall survival rate of 35%.3 Complications of bone marrow failure are a leading cause of death for these patients. The incidence of these disorders is likely to rise with the ageing of the US population making this an increasingly important public health concern.
Advanced age is the primary risk factor for developing MDS. Most patients diagnosed with MDS have no other known predisposing risks. Exposures that have been associated with subsequent development of MDS include chemotherapy, radiation, agricultural chemicals (i.e. pesticides), solvents and tobacco smoking. Of these, exposure to alkylating agent chemotherapy (i.e. melphalan) has been most well defined. Alkylating agent associated MDS typically presents within five to seven years after exposure and is characterized by abnormalities in chromosomes 5 and 7.
Diagnosis and Prognosis
Diagnosis of MDS relies primarily on peripheral blood and bone marrow findings. The diagnosis should be suspected in individuals presenting with persistent cytopenia. Careful attention should be paid to consistent decreases in blood counts over time in an older adult which may signify early developing MDS. A frequent presentation can be progressive macrocytic anaemia in an older adult. Many patients are asymptomatic at the time of diagnosis. However, careful history-taking should include questions regarding recurrent infections, bruising, bleeding, duration of cytopenia and need for red cell transfusion. The differential diagnosis for suspected MDS includes megaloblastic anaemia (B12 and folate deficiency), AML, aplastic anaemia, copper deficiency, viral infections (HIV), large granular lymphocytic leukaemia, paroxysmal nocturnal haemoglobinuria (PNH), and heavy metal poisoning.
The initial workup includes a CBC with differential, reticulocyte count, RBC folate, serum B12, iron studies and review of the peripheral smear. Classic peripheral blood findings associated with MDS include macrocytosis and hypogranular, hypolobated neutrophils (pseudo-Pelger-Huet anomaly). A bone marrow biopsy with cytogenetic analysis is necessary to confirm the diagnosis. The bone marrow is typically hypercellular with dysplastic features. The percentage of myeloblasts in the marrow helps differentiate between an MDS and AML. Cytogenetic abnormalities play an increasingly important role in the diagnosis and prognosis of MDS. Clonal chromosomal abnormalities can be detected in approximately 50% of patients with MDS.4 Diagnosis of certain MDS subtypes (such as the 5q minus syndrome) is entirely dependent upon detection of specific cytogenetic abnormalities. Cytogenetic testing, therefore, should be performed as part of bone marrow evaluation in all patients with suspected MDS to inform the diagnostic workup, prognosis and treatment options.
The classification of MDS has evolved. Historically, the French-American-British (FAB) Cooperative Group classification system was used which included five diagnostic categories based on peripheral blood and bone marrow characteristics. Categories included refractory anaemia (RA), RA with ring sideroblasts (RARS), RA with excess blasts (RAEB), RAEB in transformation (RAEBT), and chronic myelomonocytic leukaemia (CMML). A more recent classification scheme was proposed and updated by the World Health Organization (WHO) which incorporates cytogenetic abnormalities.5 This classification scheme is presented in Table 33.1 and reflects the heterogeneity of MDS.
Table 33.1 WHO Classification of peripheral blood and bone marrow findings in myelodysplastic syndromes.
| Disease | Blood findings | Bone marrow findings |
| Refractory cytopenias with unilineage dysplasia (RCUD) | Unicytopenia or bicytopenia | Dysplasia in ≤10% of cells in one myeloid lineage |
| Refractory anaemia | No or rare blasts | <5% blasts |
| Refractory neutropenia | <15% ringed sideroblasts | |
| Refractory thrombocytopenia | ||
| Refractory anaemia with ringed | Anaemia | ≥15% ringed sideroblasts |
| sideroblasts (RARS) | No blasts | Erythroid dysplasia only |
| <5% blasts | ||
| Refractory cytopenia with multilineage dysplasia (RCMD) | Cytopenias | Dysplasia in ≥10% of the cells of two or more myeloid cell lines |
| No or rare blasts (<1%) | <5% blasts in marrow | |
| No Auer rods | No Auer rods | |
| 1 × 109 l−1 monocytes | ±15% ringed sideroblasts | |
| Refractory anaemia with excess | Cytopenias | Unilineage or multilineage dysplasia |
| blasts-1 (RAEB-1) | <5% blasts | 5–9% blasts |
| No Auer rods | No Auer rods | |
| 1 × 109 l−1 monocytes | ||
| Refractory anaemia with excess blasts-2 (RAEB-2) | Cytopenias | Unilineage or multilineage dysplasia |
| 5–19% blasts | 10–19% blasts | |
| Auer rods ± | Auer rods ± | |
| 1 × 109 l−1 monocytes | ||
| Myelodysplastic syndrome-unclassified (MDS-U) | Cytopenias ≤1% blasts | Dysplasia <10% of cells in one or more myeloid cell lines accompanied by a cytogenetic abnormality that is characteristic for MDS |
| <5% blasts | ||
| MDS associated with isolated del (5q) | Anaemia | Normal to increased megakaryocytes with hypolobated nuclei |
| Usually normal or increased platelet count | <5% blasts | |
| <1% blasts | Isolated del(5q) cytogenetic abnormality | |
| No Auer rods |
Adapted from Brunning R et al. Tumours of Haematopoietic and Lymphoid Tissues. Geneva: World Health Organization, 2008.
The natural history of patients with MDS syndromes is quite variable. It is well established that mutagen-induced MDS is associated with a poor prognosis and that increased age is also a negative prognostic factor. However, the heterogeneity of this disease has complicated accurate prognostication in de novo MDS. The International Prognostic Scoring System (IPSS) (Table 33.2(a)) was developed to risk stratify patients at the time of diagnosis based on cytogenetic, morphological and clinical data. The IPSS for MDS was developed based on an analysis of 816 patients which demonstrated that specific cytogenetic abnormalities, the percentage of blasts in the bone marrow, and the number of haematopoietic lineages involved in the cytopenia were the most important variables in disease outcome.6 Risk scores are determined based on these variables, and a categorization of low risk, intermediate-1, intermediate-2, and high risk is assigned (Table 33.2(b)). The IPSS demonstrated improved prognostic discrimination over earlier classification schemes and has been incorporated into clinical practice and subsequent trial design.
Table 33.2 International Prognostic Scoring System (IPPS) for MDS.
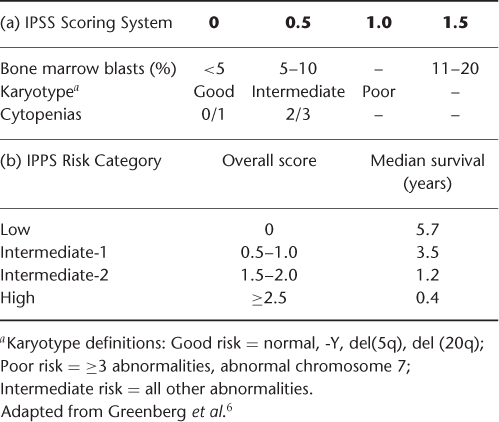
There are multiple known prognostic factors that are not included in the IPSS classification. For example, chronological age is not incorporated into the IPSS score. The prognostic impact of increasing age, however, differs by IPSS risk group. In the original analysis used to develop the IPSS, survival for high-risk patients appeared to be independent of age. This suggests survival was driven primarily by tumour biology in these patients.6 Survival for low-risk patients was strongly dependent on age with median survival of 9.0 years versus 3.9 years for age groups <70 and ≥70 years respectively. Therefore additional factors such as comorbid disease may influence survival in the setting of more indolent MDS.
The IPSS classification system has several limitations: (1) it does not account for duration of disease; (2) subjects who received prior therapy or had secondary MDS were excluded; and (3) it does not account for performance status or comorbid disease. Additional classification schemes have been proposed which include prognostic factors such as poor performance status, older age, prior transfusions and time elapsed from diagnosis.7 Despite its limitations, the IPSS classification remains a useful tool in clinical practice.
Treatment
Treatment strategies for MDS have been evolving in recent years to target higher risk MDS and subgroups defined by specific cytogenetic abnormalities. Treatment goals include managing cytopenias, decreasing progression to AML, improving overall survival while maintaining quality of life. Current treatment recommendations for MDS involve a risk-adapted therapeutic approach. In general, the National Comprehensive Cancer Center (NCCN) guidelines recommend classifying patients into relatively low risk (IPSS Low or Intermediate-1 categories) and higher risk (IPSS Intermediate-2 and High categories).8 Supportive care aimed at controlling symptoms related to cytopenias is the mainstay of treatment for: (1) low-risk patients and (2) patients with poor functional status or limiting comorbid conditions regardless of IPSS classification. Higher risk patients with good functional status and manageable comorbid conditions should be considered for low-intensity chemotherapy along with best supportive care.
Supportive care typically constitutes red cell and platelet transfusions, antibiotics for infection, haematopoietic growth factors such as recombinant erythropoietin (epoetin alpha) for selected patients, and iron chelation. Patients with symptomatic anaemia, or transfusion-dependent anaemia, may benefit from epoetin alpha or a longer acting form darbepoetin alpha. Response rates in studies are approximately 50% (major response is defined by haemoglobin increase >2 g dl−1 or transfusion independence, minor response is defined by haemoglobin increase 1–2 g dl−1 or 50% reduction for transfusion-dependent patients).9 Patients most likely to respond to use of erythropoietin are those with IPSS score low/intermediate-1, serum erythropoietin level <500, and transfusion requirement of <2 units of red cells per month.10 The NCCN recommends a target haemoglobin of ≤12 g dl−1. Granulocyte colony stimulating factor (G-CSF) may have a synergistic effect in combination with epoetin for treatment of anaemia. Low dose G-CSF dosed three times per week can be added to erythropoietin if response is inadequate to epoetin alone. Use of growth factors for symptomatic anaemia has been associated with improved quality of life and decreased transfusion requirement without increased risk of progression to AML.8, 11 However, no improvement in survival has been attributed to this intervention. Responses in haemoglobin levels are typically evident within 6–8 weeks of treatment. Ongoing treatment is not indicated if no response is detected during this time period.
Over time, most patients become transfusion dependent, increasing the risk of iron overload. Secondary iron overload negatively affects survival in transfusion-dependent patients.12 Current consensus guidelines recommend iron chelation therapy for those patients most likely to suffer negative consequences from chronic iron overload.8 The patients most likely to benefit from chelation are those with lower risk MDS, ongoing transfusion dependence, and anticipated survival of greater than one year. For these patients, initiation of iron chelation therapy should be considered after transfusion of 20–30 units of red cells or the serum ferritin exceeds 2500 μg l−1. Serum ferritin can be used to monitor efficacy of chelation with goal ferritin <1000 μg l−1 on treatment. Chelation agents include deferoxamine or the oral agent deferasirox.
Patients in the higher risk IPSS categories are more likely to experience morbidity related to cytopenias and to progress to acute leukaemia in a shorter time interval from diagnosis. For patients who present with high-risk disease or show evidence of progression to high-risk disease during follow-up, treatment with low-intensity chemotherapy should be considered. The low-intensity chemotherapeutic agents which have demonstrated efficacy in treatment of MDS are the hypomethylating agents 5-azacytidine and decitabine.
The first chemotherapy agent to demonstrate efficacy in treatment of MDS was 5-azacytidine. A randomized controlled trial of 191 patients which compared treatment with 5-azacytidine to supportive care alone for high-risk MDS demonstrated significant improvements in response, time to AML progression, and survival.13 Eligible patients (median age 68) met FAB classification criteria for MDS and were considered high risk. The 5-azacytidine was administered at a dose of 75 mg m−2 for 7 consecutive days on a 28-day cycle. The response rate (complete and partial) was 23% in the treatment arm with median time to leukaemic progression 21 versus 13 months for supportive care (p = 0.007). After controlling for the effect of crossover from placebo to active treatment, there was a survival advantage detected in treated patients.
Importantly, this study also evaluated quality of life outcomes.14 Treatment with 5-azacytidine was associated with significant improvement in fatigue, dyspnoea, self-reported physical functioning and psychological distress. Quality of life differences were maintained after controlling for the number of transfusions received. The improvements in both disease-related outcomes and quality of life established the use of 5-azacytidine as a standard of care for treatment of high-risk MDS. Another randomized controlled international trial of 358 patients confirmed an overall survival benefit favouring 5-azacytidine over physician-directed conventional care.15
Based on treatment experience from these randomized studies it is generally recommended to treat for approximately 4–6 cycles if tolerated to determine response for individual patients. Treatment protocols typically recommend treating for an additional three cycles after achieving complete remission or for as long as a treatment benefits persist in patients with a lesser response.
Decitabine, a second pyrimidine nucleoside analogue of cytidine which inhibits DNA methylation, is also FDA approved for the treatment of higher risk MDS. A randomized study of 170 patients (median age 70) compared decitabine versus supportive care for patients who met FAB classification for MDS and had an IPSS score ≥0.5.16 The dose schedule was 15 mg m−2
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


