Chapter Outline
OVERVIEW OF LYSOSOMAL STORAGE DISEASES AND THEIR DIAGNOSES
Mucolipidosis Type II/ III (I-Cell Disease and Pseudo-Hurler Polydystrophy)
Lysosomal Acid Lipase Deficiencies
Neuronal Ceroid Lipofuscinoses
This chapter provides an overview of the lysosomal storage diseases (LSDs) with particular reference to their specific hematologically related manifestations or treatments. Comprehensive reviews, such as Chapters in the Metabolic and Molecular Bases of Inherited Diseases, 9 th ed., are available for molecular genetics, biochemistry, enzymology, and phenotypes, and these sources should be consulted for details.
The Lysosomal System
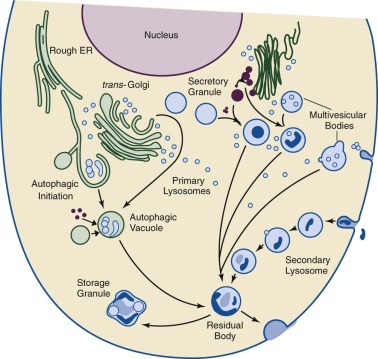
The lysosome is an intracellular organelle originally encompassing two concepts: (1) an intracellular acidic vacuole for digestion of intracellular and extracellular organic materials, and (2) biogenesis mechanisms to deliver or target its constitutive proteins, lipids, and other components. Inherent in these were the foundations of the pathogenesis and therapy for the LSDs (i.e., the lysosomal accumulation of specific substrates for particular digestive hydrolases and the potential to correct such abnormalities by the reconstitution of the appropriate levels of enzyme activity in the lysosome). These concepts, although still relevant, evolved to include the lysosome as an integral part of a major intracellular system for the clearance of internal and external macromolecules and organelles (i.e., the autophagy system). In addition, the lysosome can no longer be considered as a simple digestive vacuole but rather as a complex organelle functioning as a signal system that specifies its tissue or cellular compositions, modulates major metabolic processes (e.g., cholesterol and triglyceride synthesis), and initiates inflammation. This complexity and its integration with the autophagy system preclude the LSDs as a simple result of inert intracellular accumulations of macromolecules and require their reconceptualization to complex disorders with wide ramifications within specific cellular types and throughout the body. Although the specific accumulated substrates may have differing effects on and manifestations in selected tissues, the disruptions of intracellular clearance through autophagy and of dysregulated metabolism are generally unifying, albeit heterogeneous, concepts for the LSDs. Indeed, the LSD disruptions of the lysosomal/autophagy/mitophagy system should be more properly termed the lysosomal autophagy system diseases (LASDs) ( Fig. 25-1 ). LSD will be used here in its conventional sense, but the concept of widespread disruption of cellular processing is essential to the evolving view of these diseases. These disruptions lead to heterogeneous effects, including macrophage activation and cytokine or chemokine storms (e.g., Gaucher and Niemann-Pick diseases), dysmorphology and skeletal malformations (e.g., mucopolysaccharidoses), and neuroinflammation and neuronal apoptosis or necroptosis (e.g., neuronopathic Gaucher disease, neuronal ceroid lipofuscinosis, and Krabbe diseases).
Morphology and Physiology
Lysosomes are membrane-bound, specialized organelles that normally constitute approximately 5% to 10% of the intracellular volume. Lysosomes are found in all nucleated cells but vary in size, morphology, and content with cell type. The characteristic azurophilic-staining granules in polymorphonuclear leukocytes and eosinophils are lysosomes. Moreover, lysosomes are abundant in cells of the macrophage/monocyte system.
Ultrastructure of the lysosome reveals a unit membrane surrounding polymorphic vacuoles, which fuse with pinocytic or phagocytic vesicles containing exogenous material (heterophagy) or with components of the autophagocytic system. The undigested material remaining in secondary lysosomes becomes a residual body that can be extruded from the cell by way of an exocytosis pathway (e.g., into bile from the liver and into urine from the kidney).
Biosynthesis and Biogenesis
The lysosomal system represents the major site of intracellular compartmentalized degradation and potentially requires more than 200 different proteins for formation and modulation of its functions. However, the protein composition of the lysosome varies considerably with the tissue or cell source. The expression of these proteins, and therefore lysosomal biogenesis, has been proposed to be controlled by the master regulator, TFEB , as are many of the autophagy proteins as part of the CLEAR system for intracellular degradation of macromolecules. Lysosomes contain more than 70 hydrolytic enzymes, each with specificity to degrade selected proteins, glycoproteins, nucleic acids, polysaccharides, glycolipids, phospholipids, and neutral lipids. The lysosomal membrane contains many transmembrane- and membrane-associated proteins for maintenance of the acidic interior and its fusion capabilities.
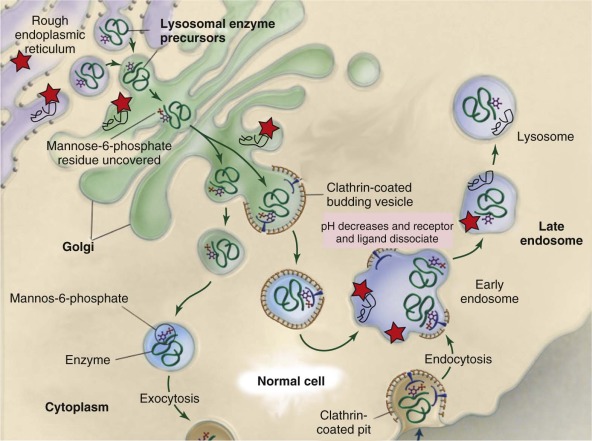
Most of the soluble lysosomal hydrolases are targeted to the lysosome by a direct route mediated by the mannose 6-phosphate receptor system. In comparison, peptide sequences, rather than carbohydrate recognition markers, target most of the lysosomal transmembrane proteins indirectly, after transport to the plasma membrane and reinternalization with delivery to the lysosome. The targeting mechanisms remain incompletely understood for several lysosomal membrane proteins, as are the roles of alternative targeting mechanisms (receptors) for some portions of soluble proteins. Many soluble lysosomal enzymes are secreted from cells, and this process forms the basis of potential cross-correction of nearby cells.
All lysosomal proteins and enzymes are synthesized on the rough endoplasmic reticulum (ER) and gain access to the ER lumen by way of a leader, or signal, peptide ( Fig. 25-2 ). The proteins are cotranslationally glycosylated on specific asparagines ( N -linked glycosylation) as they enter the ER lumen. Once glycosylated, vesicle-mediated transport delivers these proteins to the cis -Golgi. The oligosaccharide side chains are remodeled as they transit from the cis – to trans -Golgi and acquire specific sugars from Golgi glycosyltransferase that are resident in the cis , mid , and trans -Golgi. For soluble lysosomal proteins a specific trafficking signal, mannose 6-phosphate (M6P), for lysosomal localization is added in the mid-Golgi. The M6P-tagged proteins bind to the M6P receptor until they undergo an acidic pH-mediated dissociation in the late endosomes and are released into the lysosomal lumen. Small amounts of these soluble proteins are secreted from the cell and can be endocytosed by way of the M6P receptors on the plasma membrane for lysosomal delivery. This is the mechanic basis for metabolic cross-correction following hematopoietic stem cell transplantation (HSCT) in some LSDs. Membrane-bound lysosomal proteins use different receptors that recognize specific dileucine and other hydrophobic sequences for their delivery to the lysosomes. The membrane-associated enzyme, acid β-glucosidase (glucocerebrosidase; i.e., the Gaucher disease enzyme) has a unique peptide sequence that binds to LIMP-2, lysosomal integral membrane protein 2, (Grabowski, unpublished), which is the specific receptor for this enzyme. This enzyme and other membrane-bound lysosomal proteins do not normally secrete from cells and thus cannot cross-correct deficient cells.
Excellent recent reviews of lysosomal biogenesis, protein sorting, and molecular events of the dynamic lysosomal and autophagy systems are available, and the reader is referred to these for details.
Pathogenesis
Hers conceptualized the inborn lysosomal diseases in 1965 and proposed that excessive lysosomal accumulation of substrate leads to an increased number of lysosomes within cells, disruption of normal cell functions, and possibly cell death. In general, the signs and symptoms of each LSD primarily reflect the normal pattern of distribution of the enzymes’ substrates. In Gaucher disease type 1 (discussed later), turnover of senescent erythrocyte and leukocyte membranes containing glucosylceramide leads to its accumulation in the macrophage/monocyte system. Similarly, disorders of ganglioside metabolism involve principally the central nervous system (CNS) because gangliosides normally have higher turnover in this tissue. Compounds that are normally abundant in all tissues lead to generalized organ dysfunction (e.g., mucopolysaccharidoses and glycoproteinoses). Because of the disruptions of the lysosomal and autophagy system, secondary accumulations of various compounds do occur; the mechanisms for such accumulations are poorly understood.
Overview of Lysosomal Storage Diseases and Their Diagnoses
The LSDs are caused by deficiencies of a single or multiple lysosomal proteins. This underscores the lack of redundancy for these proteins and their importance in maintaining cell homeostasis. Most LSDs result from single enzyme or protein deficiencies resulting from mutations in their respective genes. The essential need for specific enzymes implies that if a single enzyme is deficient or functionally defective, the hydrolytic pathway is halted at the level of the missing enzyme, and its specific substrate (or substrates) accumulates in the lysosome with resultant organelle, cellular, and organ dysfunction. Some LSDs have multiple enzyme or protein deficiencies that result from mutations in single genes that are necessary for posttranslational processing or assembly or stability of a multiprotein complex. Examples include mucolipidosis II and III that disrupt the M6P targeting of many soluble lysosomal proteins to the lysosome, thereby leading to intracellular deficiencies of most soluble lysosomal enzymes. Multiple lysosomal hydrolase defects also result from single gene defects that alter specialized posttranslational processing or multienzyme complex formation. Multiple sulfatase deficiency, in which all, not only lysosomal, cellular sulfatase activities are deficient, results from a single gene defect that encodes an active site modification enzyme that is essential for the activity of all sulfatases. Multiprotein complex formation is defective in combined β-galactosidase/neuraminidase/“protector protein” deficiency in which there is a deficiency of a “protective protein (cathepsin A)” that is essential for the formation and stability of the tri-enzyme complex.
All LSDs are inherited in an autosomal recessive manner, except for Fabry disease, Hunter syndrome (MPS II), and Danon disease, which are X-linked. Extensive allelic heterogeneity exists in all the LSDs, including many single base substitutions, premature chain terminations, and insertions and deletions of part or all of the respective genes. Some of the LSDs exhibit founder effects leading to a common mutation (or mutations) in specific demographic or ethnic groups. New alleles are regularly being discovered in all of the LSDs, and databases for many LSD mutations are maintained. Polymorphic variation also occurs in some of the alleles, and these can affect the resultant phenotype of a specific disease-causing genotype. Imperfect genotype-phenotype correlations exist, but in general this information can inform the anticipated prognosis and therapeutic options (discussed later).
Because many of the enzyme-defective LSDs are very rare, enzyme assays remain the gold standard for antenatal and postnatal diagnoses. Indeed, identification of a “mutation of unknown clinical significance,” which often occurs in unrelated families with an LSD, requires enzyme confirmation of the diagnosis. For known pathogenic mutations in genes encoding enzymes and proteins in specific families or ethnic groups, molecular diagnosis is highly reliable, particularly for heterozygote detection and prenatal diagnosis.
For enzyme diagnosis, serum, plasma, or mixed leukocytes provide readily available sources, and reliable assays using synthetic fluorogenic or colorimetric substrates are available. LSD enzymes require different sources for assay; for example, serum and plasma or peripheral blood leukocytes are routinely used for many of the LSD-associated enzyme assays. Nucleated cells are needed for the diagnosis of Gaucher disease because the relevant enzyme is not secreted. Recently, dried blood spots have become more generally available for screening studies, but definitive enzyme assays should be used for confirmation. Antenatal diagnosis is available for most LSDs. Such sample needs are specified on the website Gene Tests (ncbi.nlm.nih.gov/sites/GeneTests/) .
The Sphingolipidoses
The sphingolipidoses are characterized by defective or absent lysosomal hydrolysis and accumulations of specific sphingolipids in lysosomes. Mutations leading to either defective or deficient function of sphingolipid hydrolases or their specific activator proteins (also known as saposins ) are the bases for these diseases. The shared structures of the sphingolipids include sphingosyl and fatty acyl acid chains that are covalently linked to form the ceramide. Attachment of various head groups to the ceramide backbone produce sphingomyelin, a phospholipid, and the more than 400 glycosphingolipids that contain single to multiple glycosyl residues. The glycosyl, fatty acid acyl chain, and sphingosyl components vary in structure with tissue, cellular, and subcellular localization. The sphingolipids are crucial structural elements of cell membranes and have important roles in cell signaling and recognition. Among the sphingolipidoses, Gaucher disease; Niemann-Pick disease types A, B, and C; Farber disease; GM 1 gangliosidosis; and GM 2 gangliosidosis have significant hematologic manifestations and effects.
Gaucher Disease
Gaucher disease, one of the more common LSDs, is an autosomal recessive trait resulting from more than 350 mutations at the GBA1 locus and the resultant deficient or defective activity of the acid β–glucosidase (glucocerebrosidase). Gaucher disease has a frequency of approximately 1/57,000 in the general population, and the type 1 variant has its greatest frequency in the Ashkenazi Jewish population, with a frequency of approximately 1/855.
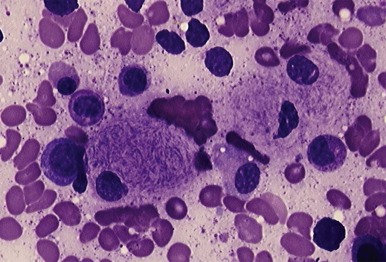
Decreased catalytic activity or instability of acid β–glucosidase leads to accumulation of glucocerebroside (glucosylceramide) and its deacylated analog, glucosylsphingosine, in the lysosomes of visceral cells of the monocyte lineage, termed Gaucher cells. Gaucher cells ( Fig. 25-3 ) accumulate in the liver, spleen, bone marrow, lung, and lymph nodes, leading to hepatomegaly, splenomegaly, anemia, thrombocytopenia, destructive bone disease, bone marrow infiltration, pulmonary disease, and lymphadenopathy.
Three clinical variants of Gaucher disease are classically described, depending on the absence or presence and severity of primary CNS involvement. Gaucher disease type 1, the “non-neuronopathic” variant, accounts for approximately 90% of all cases in European and North American populations. Type 1 disease displays marked phenotypic variability in its age at onset, rate of progression, and degree of visceral organ involvement. Although survival may be reduced, individuals may live well into adulthood. Indeed, patients have been diagnosed coincidentally in the sixth to eighth decades of life, and with particular genotypes the manifestations may be so minor that affected patients do not seek medical attention.
Gaucher disease types 2 and 3 are primary neuronopathic variants distinguished by the age of onset and severity of CNS features. Although infrequent in European and North American populations, they are more commonly seen in Asia, Africa, and South and Central America. The demarcation between Gaucher disease types 2 and 3 is clinically useful but somewhat arbitrary insofar as these variants represent a continuum of disease phenotypes. Gaucher disease type 2 is a severe, early (prenatal to first 6 months of life) onset, rapidly progressive neurologic disease, particularly with brainstem abnormalities (stridor, dysphagia, aspiration) and visceral disease. Death occurs in the first 1 to 2 years of life. Gaucher disease type 3 has a more protracted course, with variable CNS and visceral organ (e.g., hepatosplenomegaly, poor weight gain) involvement. Indeed, horizontal supranuclear gaze palsy and minor cognitive deficits may be the only manifestations of neurologic disease for decades. Survival is shortened, with death usually occurring by the fourth decade of life.
Although Gaucher disease type 2 demonstrates similar visceral findings to types 1 and 3, only the findings in Gaucher disease type 1 are presented here. The visceral findings in types 1 and 3 are very similar. It is important to note that types 1 and 3 can present in the first few years, and therefore the distinction between types 1 and 3 is not based on age at onset but rather by the presence of early primary CNS involvement in type 3.
Hematologic abnormalities are common in individuals with Gaucher disease type 1. Thrombocytopenia of mild to moderate severity is the most common finding in the peripheral blood and is seen in 50% to 70% of individuals at diagnosis. Splenic sequestration and diminished platelet production caused by bone marrow infiltration are implicated as mechanisms. Splenectomy improves platelet counts; however, this is generally not advised because of the associated risks, as described later.
Anemia is present in approximately 20% to 30% of clinically involved individuals at diagnosis, although this varies with GBA1 genotype. The anemia is generally mild and is normocytic or microcytic and hypochromic. In developing countries severe anemia in individuals with Gaucher disease may be multifactorial, and additional contributing factors should be sought. Low serum iron levels and iron-binding capacity resemble the anemia of chronic disease, but excessive iron is present in bone marrow Gaucher cells. Ferritin levels are generally elevated, likely owing to the inflammatory component of Gaucher disease. Iron therapy is generally not useful, but a short trial of iron may provide insight into its utility. Megaloblastic anemia occurs with vitamin B 12 deficiency, which may be more frequent in this disease; evaluation for vitamin B 12 deficiency should be considered, with appropriate treatment as indicated. Anemia requiring blood transfusions is unusual in developed countries.
Coagulation factor abnormalities are common, including abnormalities of factor IX, vWF Ag, and factor VIII. These rarely require clinical intervention and may be incidental findings. Deficiencies of factor XI are frequent, particularly in the Ashkenazi population.
Leukopenia is atypical but may occur with severe splenomegaly or bone marrow failure caused by myelofibrosis. Recently, the roles of excessive cytokine and chemokine have been recognized as important to the pathology of Gaucher disease. This is because of the recruitment and chronic stimulation of immune system cells, including B- and T-lymphocytes. The long-term stimulation of B-cells and their precursors lead to monoclonal and oligoclonal gammaglobulinemias and an increased rate of multiple myeloma in adults. Autoimmune hemolytic anemia occurs infrequently
Most patients (75% to 90%) have bone abnormalities on radiographic imaging. At least half have an Erlenmeyer-flask deformity, in which expansion of the femoral midshaft leads to a tapered appearance and widening of the distal end. Destruction of the marrow cavity (resulting from marrow replacement by Gaucher cells), cortical lytic lesions (caused by infarctions), and osteopenia or osteoporosis are present in many patients. Painful bone crises, similar in clinical manifestations to bone infarcts in sickle cell disease, occur in approximately 10% of patients and are more frequent in the femoral head and shaft. These episodes can mimic osteomyelitis, with fever, local erythema, leukocytosis, and increased erythrocyte sedimentation rate. Growth retardation occurs in 35% to 40% of children, leading to a reduction in predicted adult height.
Hepatomegaly is nearly universal in Gaucher disease types 1 and 3, although the degree of enlargement is variable, ranging from 1.25 to greater than eightfold of predicted normal volumes. The liver is usually homogenous, with evidence of fatty infiltration on magnetic resonance imaging (MRI) studies, but isolated areas of Gaucher cell accumulations do occur and may raise the suspicion of malignancy. Persistent mild elevations in serum transaminases and increased unconjugated bilirubin are not infrequent; however, liver failure is rare and is associated with other comorbidities, including hepatitis C. Hepatocellular carcinomas and cholangiosarcomas have occurred, but these are unusual and confined to the adult population.
Asymptomatic splenomegaly is the most frequent presenting sign. Splenomegaly may be sufficiently pronounced to result in early satiety or to interfere mechanically with respiratory function. As evaluated by MRI or ultrasound, the spleen is generally homogeneously enlarged early in life. With increasing size and age, the spleen can develop infarctions, multiple areas of scarring, and intrasplenic nodules. The scarred or damaged areas may lead to intrasplenic consumption of coagulation factors. In adults malignancy should be excluded. Although infrequent, pain from splenic infarction may mimic an acute abdomen. Splenectomy rapidly improves the hematologic signs, but it is generally not recommended because it has been linked to worsening bone disease and increased frequency of pulmonary hypertension.
Several variants of pulmonary involvement occur in Gaucher disease type 1, but they are much more frequent in type 3. These include interstitial infiltrative disease, alveolar infiltration, hepatopulmonary syndrome, and pulmonary hypertension and are associated with significant morbidity and mortality risk. The infiltrative diseases are more common in younger patients with type 3 disease and can be exacerbated by chronic aspiration. Pulmonary vascular disease is associated with adult female, non-Jewish patients who have undergone splenectomy. These complications can be challenging to treat and require medications for pulmonary hypertension as they respond poorly to enzyme therapy.
Specific malignancies have a clearly increased frequency in adults with Gaucher disease type 1. These include multiple myeloma and lymphoid cancers (i.e., leukemia, Hodgkin and non-Hodgkin lymphoma). The lifetime risk of multiple myeloma in untreated type 1 disease is estimated to be approximately 25 times that of the general population. The overall cancer risk in patients with Gaucher disease is estimated at 1.8 times that of the general population. However, the significance of this and the relationship to Gaucher disease is unclear. There are no data to support an increased risk of malignancy in children with types 1 and 3 diseases.
Gaucher cells are the hallmark histologic findings in all variants of Gaucher disease. These lipid-laden macrophages are approximately 20 to 100 µm in diameter, with tubular inclusions resembling crumpled tissue paper (see Fig. 25-3 ). This material stains positively with periodic acid–Schiff (PAS) stain. In comparison, storage cells in sphingomyelinase deficiency (discussed later) contain foamy-appearing inclusions. On electron microscopy Gaucher cells demonstrate membrane-bound inclusions containing twisted structures ( Fig. 25-4 ). Histologic analysis of bone marrow demonstrates accumulation of Gaucher cells and fibrosis; the latter can be progressive.
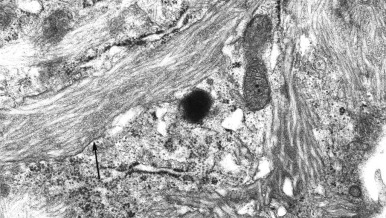
Clinical features suggest the diagnosis of Gaucher disease, as does the presence of Gaucher cells in tissue (i.e. bone marrow, liver, spleen) or bronchoalveolar lavage. Confirmatory laboratory diagnosis requires demonstration of decreased acid β-glucosidase activity in nucleated cells or tissues, most commonly peripheral blood leukocytes or cultured skin fibroblasts. Molecular diagnosis through the identification of pathogenic mutations in both copies of GBA1 can provide useful prognostic information. Although genotype and phenotype correlations are not absolute, identification of known mutations may provide insight into the potential severity and risk of CNS or non-CNS disease. For example, the presence of a GBA1 mutation encoding N370S in affected individuals is not associated with early onset progressive, primary neurologic disease. Homozygosity for N370S generally predicts a less severe disease course than other genotypes. About 50% of N370S homozygotes are predicted never to come to medical attention owing to their lack of overt signs and symptoms. For example, N370S homozygotes have an age of onset or recognition of about 32 years, whereas in individuals with the N370S/84GG (null), onset occurs by approximately 6 years. In addition, the latter genotype is associated with greater liver and spleen enlargement. Homozgyosity for the recurrent allele encoding L444P is highly associated with early onset primary CNS disease. An extensive review of these correlations from large patient populations is available.
Recommendations from expert panels regarding evaluation, monitoring, and treatment of patients with Gaucher disease are available and revised on a regular basis. Gaucher disease variants are multisystem diseases that affect disease management plans. The lack of predictive biomarkers necessitates comprehensive, multidisciplinary evaluations of the major organs for associated complications. The primary focus is on the major involved organs (liver, spleen, blood, and bone) and their functions.
Although HSCT has been used to treat type 1 disease, its utility is limited by the availability of safer, specific forms of therapy. Because acid β-glucosidase is not normally secreted from cells, HSCT has not shown significant or consistent effects on the CNS manifestations in type 3 disease. Similarly, HSCT in type 2 disease has not been successful for the CNS disease.
Two treatment approaches are currently available for the treatment of Gaucher disease type 1: enzyme replacement therapy (ERT) and substrate reduction therapy (SRT). With ERT recombinant human acid β-glucosidase, a glycoprotein, is administered intravenously. For Gaucher disease the available ERT products have been modified to expose terminal α-mannosyl residues for preferential targeting to the macrophage mannose receptor. After intravenous administration such modified enzymes preferentially localize to the macrophage compartment, are internalized by receptor-mediated endocytosis, and delivered to the lysosomes where they reconstitute the enzyme activity. This leads to degradation of the stored glucocerebroside and other glucolipids. SRT is different and depends on the presence of some residual mutant enzyme activity (1% to 15%) that can degrade a lesser amount of substrate and establish a healthier steady state. The driving concept is to slow the synthesis of the offending substrates by inhibition of their synthesis. The target is the Golgi localized enzyme, glucosylceramide synthase. For either ERT or SRT, the efficacy will be limited by tissue delivery and penetration. For example, the ERT recombinant enzymes are macromolecules that do not penetrate the blood-brain barrier, or potentially other tissue barriers, in sufficient quantities to be therapeutic. The SRT small molecules may or may not be distributed to all tissue and can have off-target effects.
ERT is currently the standard of care for the treatment of Gaucher disease. Three ERT products are currently approved: Imiglucerase (Cerezyme, Genzyme, a Sanofi company, Cambridge, Mass., approved by the Food and Drug Administration [FDA] in 1991), velaglucerase alfa (VPRIV, Shire Human Genetic Therapies, Inc., Cambridge, Mass., approved by the FDA in 2010), and taliglucerase alfa (Elelyso, Pfizer, Inc., New York, N.Y., approved by the FDA in 2012). The FDA currently restricts the use of taliglucerase alfa to adults (older than 18 years). These products differ in their manufacturing processes, glycosylation patterns, and amino acid sequence. Briefly, they are all human acid β-glucosidases that are produced in large cultures of Chinese hamster ovary cells (imiglucerase), human fibrosarcoma cells (velaglucerase alfa), or carrot root cells (taliglucerase alfa). All have terminal α-mannosyl residues and target the macrophage mannose receptor; have highly similar crystal structures; and have similar, if not identical, enzyme functions. On the basis of current data, all three ERT products have similar clinical effects and safety. Direct head-to-head studies of imiglucerase and velaglucerase alfa were conducted in mouse analogs of Gaucher disease and shown to have similar biochemical, histopathologic, and antibody formation effects.
The long-term (approximately 20 years) safety and efficacy of imiglucerase has been shown in Gaucher disease type 1. In 1028 such patients, hemoglobin concentrations increased to normal or near normal within 6 to 12 months, with a sustained response through 5 years. In thrombocytopenic patients with intact spleens, improvements in platelet counts were particularly noted during the first 2 years of therapy. Further increases in platelet counts were observed during years 2 through 5; The likelihood of achieving a normal platelet count decreased with increasing severity of baseline thrombocytopenia. Similarly, significant decreases in liver (20% to 30%) and spleen (30% to 50%) volumes were noted after 2 years, with additional reductions in liver volume to near normal levels by 5 years. Further decreases in spleen volume after two years were minimal. Improvement or stabilization of bone pain occurred in many patients after 2 years of therapy, with approximately a third of patients having residual bone pain reports, probably the result of chronic joint abnormalities that are not directly related to Gaucher disease. A 10-year follow-up of patients treated with imiglucerase showed sustained stability of disease. The safety profile of imiglucerase has been extensively reviewed and shows that the drug is generally safe and well tolerated. A detailed statistical study of various doses of imiglucerase shows a clear dose response of the major monitoring parameters and provides guidance for expected responses in patients initiated on any dose between 15 to 60 U/kg every two weeks. Bone mineral density was also dose dependent over the long term. About 13% of patients receiving ERT with imiglucerase develop immunoglobulin G (IgG) antibodies, and about half of these patients have minor adverse events or infusion-related events. These are generally easily controlled. Inhibitor antibodies have been reported in a few patients, and immunoglobulin E (IgE) antibodies have been anecdotally reported in fewer than 10 patients. Similar long-term data are not available for velaglucerase alfa and taliglucerase alfa, but phase II and phase III studies show comparable safety and efficacy profiles to imiglucerase.
SRT with miglustat (Zavesca, Actelion Pharmaceuticals, South San Francisco, Calif.) is licensed in several countries for use in adults with mild to moderate Gaucher disease type 1 for whom ERT is unavailable or contraindicated. Miglustat is an oral medication that produces statistically significant effects on liver and spleen volumes but only slight improvements in hematologic variables. Miglustat is reported to positively affect bone mineral density. The drug has several side effects, including diarrhea (approximately 80%) and peripheral neuropathy, which may be significant in some patients.
Phase I and phase II clinical studies on a second SRT, eliglustat tartrate (Genzyme, a Sanofi company; Cambridge, Mass.), have been completed, and initial phase III results are available. In the phase II study, 20 patients completed a total of 2 years of therapy. In these patients statistically significant improvements from baseline were observed for platelet count, hemoglobin level, spleen volume, liver volume, and bone mineral density of the lumbar spine. These findings were essentially duplicated in the initial phase III data in patients who were naïve to therapy. Also, the drug was well tolerated, without many of the side effects observed in miglustat.
Acid Sphingomyelinase Deficiency (Niemann-Pick Disease Types A and B)
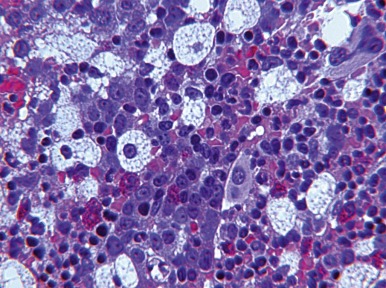
The acid sphingomyelinase (ASM) deficiencies are a group of clinically heterogeneous, autosomal recessive conditions, characterized by deficient activity of ASM resulting from mutations in SMPD1 . Sphingomyelin, a phosphosphingolipid, accumulates in lysosomes of multiple cell types, particularly those of the macrophage or monocyte lineage, leading to characteristic lipid-laden Niemann-Pick foam cells ( Fig. 25-5 ).
Two specific variants of ASM deficiency are classically recognized. Niemann-Pick A (NPA) is an early onset neurologic disorder characterized by progressive psychomotor retardation, ocular findings (cherry-red spots [approximately 50%]), massive hepatosplenomegaly, and cachexia, leading to death in the first few years. A nonneurologic form (NPB) involves visceral disease and is compatible with survival into adulthood. The NPB phenotype includes growth retardation, hepatosplenomegaly, infiltrative pulmonary disease, pancytopenia, skeletal disease, hypercholesterolemia, and lymphadenopathy. Intermediate phenotypes between NPA and NPB are frequently described. As with many LSDs, ASM deficiencies represent a disease continuum.
The incidence of the various forms of the disease varies among different populations. Based on long-term experience in Australia, the estimated combined frequencies of NPA and NPB are approximately 1 in 248,000. NPA is more prevalent in the Ashkenazi Jewish population, in which approximately 1 in 80 persons are heterozygotes. NPB is panethnic, with the highest incidence in individuals of Middle Eastern and North African descent. Emerging data suggest greater frequencies in Brazil, but better ascertainment is needed.
NPA presents somewhat uniformly and includes severe progressive neurovisceral disease within the first few months of life. Neonatal jaundice, hepatomegaly (usually in the absence of liver dysfunction), splenomegaly, and failure to thrive are present early. Moderate lymphadenopathy and infiltrative pulmonary disease are frequently seen. Moderate microcytic anemia, which may respond to iron supplementation, and thrombocytopenia develop progressively. During the subsequent months neurodevelopmental arrest is followed by regression, hypotonia, muscle weakness, and ultimately spasticity and rigidity as terminal stages. Ophthalmologic examination may reveal cherry-red maculae in approximately 50% of subjects. Death usually occurs by 2 to 3 years of age.
NPB is a heterogeneous disease with manifestations generally restricted to the visceral organs. Most patients are diagnosed in childhood, often after hepatosplenomegaly is found on routine examination. Hepatosplenomegaly is most prevalent during childhood and less so in adults. The mean liver and spleen volumes are 1.9±0.7 MN [range: 0.7-3.9 MN (MN=calculated multiples of normal)] and 11.1±5.7 MN (range 3.1-27.3 MN), respectively. About 71% of individuals had liver volumes greater than 1.5 MN, whereas 84% of individuals had spleen volumes greater than 5 MN. Splenectomy has been performed in the past, particularly in those with a history of bleeding and thrombocytopenia. The removed spleens are bright orange as a result of the sphingomyelin accumulation. Splenectomy is generally discouraged because of concerns that disease severity may worsen in other organs, such as the lung, and the risk of infection related to splenectomy.
Atherogenic serum cholesterol and lipoprotein profiles include low high-density lipoprotein (HDL), and elevated total cholesterol, triglycerides, low-density lipoprotein (LDL) and very-low-density lipoprotein (VLDL) levels. These are present from early childhood. Additionally, easy bruising and bleeding, infiltrative pulmonary disease, and joint or limb pain (or both) are common. Some individuals have significant bleeding (i.e., epistaxis, postsurgical bleeding) and may require medical intervention, including surgical procedures and blood transfusions. Thrombocytopenia is the most common hematologic finding, followed by anemia and leukopenia. These hematologic abnormalities are generally mild to moderate. In a cross-sectional study of patients with NPB, mean hemoglobin and hematocrit concentrations were 13.3 g/L (SD 1.5; range 9.3-16.5) and 39.1% (SD 4.5; range 27.8-48.3), respectively, whereas mean white blood cell (WBC) and platelet counts were 6.4 × 109/L (SD 2.7; range 2.1-16.2) and 158 × 109/L (SD 82; range 59-459). Osteoporosis, expansion and widening of the medullary cavities, and remodeling defects can be demonstrated radiologically. Survival into adulthood is common, as is development of liver cirrhosis and fibrosis and pulmonary dysfunction.
Although NPB is not generally associated with primary neurologic involvement, a subset of such patients may have exhibit retinal abnormalities, including cherry-red maculae or a gray, granular pigmentation around the fovea. Cognitive impairment has been found with or without other neurologic findings. These findings highlight the continuum of Niemann-Pick disease variants, rather than discrete subtypes.
Niemann-Pick cells, a primary histologic finding in ASM deficiencies, are approximately 25 to 75 µm in diameter. The cytoplasm is filled with lipid vacuoles, imparting a foamy appearance to the cells ( Fig. 25-5 ). The vacuoles stain positive with Sudan black B and oil red O but poorly with PAS. Within the bone marrow the intracellular material in Niemann-Pick cells may have a bluish color on Giemsa staining and are occasionally referred to as sea-blue histiocytes . Sphingomyelin also accumulates in hepatocytes, with inflammation and fibrosis that can lead to cirrhosis ( Fig. 25-6 ). Alveolar histocyte-like cells filled with sphingomyelin accumulate in the lung.

ASM deficiency is suspected on the basis of the clinical features and identification of Niemann-Pick cells on histologic examination of tissues. The diagnosis is confirmed by deficient ASM activity in any nucleated cell sample or identification of pathogenic mutations in both copies of SMPD1 . Of note, the type and severity of disease correlate poorly with the amount of in vitro residual enzyme activity.
No specific treatments for NPA or NPB are currently approved. Treatment is supportive. HSCT has been performed in patients with NPA and NPB. Despite engraftment, continued neurologic deterioration occurs in NPA. HSCT has been disappointing in NPB. Variable effects have been seen on the liver and lung manifestations. Two well-documented cases are instructive. Both showed improvement or resolution of pulmonary disease but no correction of hepatocellular disease despite resolution of Kupffer cell involvement. Indeed, both patients progressed to cirrhosis. A phase I clinical trial evaluating the safety of ERT in NPB adults has been completed, and a phase II trial is currently being conducted.
Niemann-Pick Disease Type C
Niemann-Pick disease type C (NPC) is a clinically heterogeneous progressive neurovisceral disease resulting from mutations of NPC1 (~95% of cases) or NPC2 (~5% of cases) and is inherited in an autosomal recessive manner. Although the major phenotypic manifestations are common to both NPC1 and NPC2, severe pulmonary disease is a unique complication and a significant source of morbidity in patients with NPC2 mutations. NPC was originally thought to be part of the NPA and NPB family, but the identification of the molecular lesions and the role of NPC proteins in cholesterol metabolism revealed a different underlying biology. However, the nosology is prevalent in the literature and has been retained.
NPC, which is panethnic with a frequency of approximately 1 in 120,000 to 1 in 150,000, is characterized by defective lipid transport and lysosomal accumulation of lipids (unesterified cholesterol, phospholipids, sphingomyelin, and glycolipids) in affected cells. The gene products of NPC1 and NPC2 are lysosomal proteins that are critical to the egress of cholesterol from lysosomes for the modulation of cellular lipid metabolism by way of the SREBP system. Although partial deficiencies of ASM activity have been found in NPC patient cells, this is not the primary defect.
NPC has three variants, based on the age of onset, severity of CNS disease, and survival: neonatal/infantile, childhood, and adolescent/adult. As with many LSDs, overlap exists among the various clinical forms, thereby forming a continuum of overlapping disease phenotypes.
Progressive neurologic or psychiatric manifestations and visceral (liver, spleen, and potentially lung) involvement occur in any of the variants. It is important to note that visceral findings may occur before or after the development of neurologic symptoms, and in adults visceral findings may be minimal at diagnosis. The age of onset of systemic involvement does not correlate with neurologic involvement. The age of onset of neurologic disease correlates with disease severity and survival.
The neonatal and infantile variant is a rapidly progressive neurovisceral disease with onset in the first few months of life and accounts for approximately 20% to 30% of cases. The most severe cases may present perinatally with ascites or hydrops fetalis ( Table 25-1 ). Infantile cholestatic disease or idiopathic neonatal hepatitis often develops in early infancy and is a common cause of neonatal liver disease. In approximately 10% of cases, fulminant liver disease develops and can lead to death. Survivors usually experience resolution of severe liver disease by 2 to 4 months of life; however, hepatosplenomegaly often remains and becomes less conspicuous with age. Failure to thrive and growth retardation are prominent. A subset of NPC2 individuals develops significant pulmonary disease, characterized by tachypnea, infiltrates on chest x-ray, and a presentation resembling alveolar proteinosis.
| Diagnosis | Fetal Features | Neonatal Findings | Pathology | Diagnostic Confirmation |
|---|---|---|---|---|
| Morquio IV A | Ascites, pleural effusion | May resolve by term | Reduced N -acetylgalactosamine-6-sulfatase activity | |
| Mucopolysaccharidosis VII | Nuchal thickening, ascites | Coarse features | Vacuoles in lymphocytes and placenta | Reduced β-glucuronidase activity |
| Gaucher | Hydrops, hepatomegaly, fetal hypokinesia | Collodion-like skin, contractures, hepatosplenomegaly | Gaucher cells | Reduced acid β-glucosidase activity |
| Niemann-Pick A | Nuchal thickening, hydrops | Hepatomegaly, prolonged jaundice, poor feeding | Foam cells | Reduced sphingomyelinase activity |
| Niemann-Pick C | ascites | Prolonged jaundice, pulmonary infiltrates | Foam cells | Abnormal filipin staining, molecular testing for NPC1 and NPC2 |
| GM 1 gangliosidosis | Intrauterine growth retardation, oligohydramnios | Hypertonia, periorbital edema, cherry red spots | Placental vacuolization | Decreased β-galactosidase, normal neuraminidase activities |
| Galactosialidosis | Hydrops | Edema, coarse facies, hepatosplenomegaly, telangiectasias | Vacuoles in circulating cells, Kupffer cells, other organs | Decrease in both β-galactosidase and neuraminidase activities |
| Infantile sialic acid storage disease | Ascites, IUGR, nuchal thickening | Hepatomegaly, coarse facies, hypotonia, stippled epiphyses | Placental vacuolization | Increased free sialic acid in urine, amniocytes, fibroblasts |
| Mucolipidosis II | Short femurs | Gingival hyperplasia, coarse facies, periorbital puffiness, hepatosplenomegaly | Vacuoles in lymphocytes, not present in bone marrow | Increase in various lysosomal enzymes in plasma |
Those recovering from early liver disease experience progressive neurodegenerative and exhibit hypotonia, developmental delay, regression, spasticity, dystonia, dysphagia, seizures, and pyramidal signs. Vertical supranuclear gaze palsy is a classic finding for NPC variants. Death usually occurs by 5 years.
Childhood-onset NPC is the most common variant, accounting for approximately 50% to 70% of cases. These children initially appear healthy, with normal development. By early to late childhood, they exhibit neurologic deterioration manifested initially as clumsiness and gait abnormalities, behavioral problems, poor academic performance, and supranuclear gaze palsy. Gradual progression of neurodegenerative signs occurs, including psychiatric disturbances, ataxia, dystonia, seizures, cataplexy, dysarthria, and dysphagia. Individuals eventually become incapacitated. Death usually occurs in the second or third decade of life, usually from pneumonia. Significant liver dysfunction is unusual in this variant.
The adolescent- or adult-onset variant accounts for approximately 5% to 10% of cases. This variant manifests primarily as a psychiatric or neurocognitive degenerative disease in which significant visceral involvement is unusual, although hepatosplenomegaly may occur. Hematologic abnormalities are not commonly present. Vertical supranuclear gaze palsy may be subtle and difficult to recognize.
Histologic analysis of bone marrow reveals characteristic foam cells containing accumulated free cholesterol, as evidenced by filipin positivity, and sphingolipids ( Fig. 25-7 ). Sea-blue histiocytes may be present. These cells are also present in other visceral organs (hepatocytes in the liver) and CNS (neurons and glial cells). Ultrastructurally, the storage material consists of pleomorphic membrane-bound lipid inclusions with structures varying from crystalline to electron-dense laminated inclusions. ,
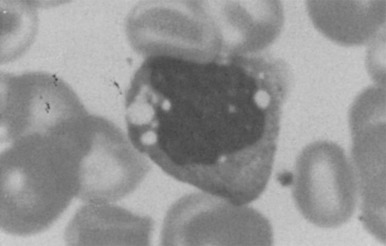
Because of the significant variability of phenotypes, NPC is not always initially considered in the differential diagnosis. Indeed, the diagnosis may be delayed as a result of the absence of hepatosplenomegaly or storage cells in the bone marrow. A biochemical diagnosis is established by demonstration of impaired cholesterol esterification and positive filipin staining in cultured fibroblasts. ASM activity is normal to low. Molecular studies can provide a definitive diagnosis; Many NPC1 or NPC2 mutations are confined to single families.
Treatment of NPC is supportive. Bone marrow transplantation may improve visceral disease but is ineffective for CNS disease. As the NPC1 protein is an integral lysosomal membrane protein with multiple transmembrane domains, it is not secreted and therefore cannot cross-correct neighboring cells. For similar reasons, liver transplantation does not benefit patients with CNS disease.
A number of pharmacologic agents have been evaluated as potential therapies for NPC, including cholesterol-lowering medications (statins), sterol-binding agents, and glucocorticoids, all without success. The safety and efficacy of miglustat (Zavesca ® , Actelion Pharmaceuticals) has been evaluated in NPC1 and may stabilize oculomotor manifestations, dysphagia, and ambulation deficits. However, the effects on visceral disease manifestations in NPC were not significant. Side effects were similar to those discussed previously and included diarrhea, flatulence, and abdominal pain. The European Medicines Agency (EMA) approved Miglustat in 2009 for the treatment of CNS manifestations in NPC. It is not approved by the FDA.
Cyclodextrin, a cholesterol-binding excipient, is being explored as a therapy for NPC. This agent is theorized to reverse the transport defect in the late endosomal or lysosomal complex, allowing sequestered free cholesterol to be transported out of the lysosome and metabolized by other cellular mechanisms. Based on encouraging preclinical studies in the Npc1 -/- mouse model, clinical trials in humans have begun.
Farber Disease (Lipogranulomatosis)
Farber disease is a very rare progressive autosomal recessive disease resulting from deficient activity of acid ceramidase, which is encoded by ASAH1 . The frequency is estimated to be less than 1 in 1,000,000. Seven subtypes of Farber disease are recognized; these vary in severity and survival. Types 6 and 7 are not caused by mutations in ASAH1 and will not be discussed here.
Classic Farber disease, the most common variant, is characterized by a triad of progressive clinical manifestations: painful and progressive deformation of joints; subcutaneous nodules, particularly near the joints and over pressure points; and progressive hoarseness resulting from laryngeal nodules. Often these findings are present within the first few weeks to months. Progressive respiratory and swallowing difficulties result in poor weight gain and impaired acquisition of development milestones; this can necessitate palliative management (i.e., tracheostomy and gastrostomy tube placement). Granulomatous lesions may also occur in the liver, spleen, lung, heart, reticuloendothelial system, bone, and CNS and contribute to the morbid manifestations. Hepatomegaly may be identified in patients. Death usually occurs within the first years of life.
Intermediate and attenuated variants demonstrate similar, albeit less severe, phenotypes. Clinical manifestations begin within the first 1 to 2 years; some individuals have survived into adulthood.
The neonatal-visceral variant is characterized by hepatosplenomegaly first noted in the neonatal period. In the most severe cases, patients may present with nonimmune hydrops fetalis. Infants with this severe phenotype die in early infancy, often before the onset of the more characteristic clinical features of classic Farber disease.
The neurologic form is defined by progressive neurodegeneration noted within the first two years, followed by death shortly thereafter. Visceral organ involvement, including subcutaneous nodules and joint involvement, may be present but does not predominate.
The principal pathologic findings are granulomatous infiltrates composed of foam cells containing a PAS-positive material that is extractable with lipid solvents. Comma-shaped curvilinear tubular, membrane-bound structures (Farber bodies) ( Fig. 25-8 ) within vacuoles are characteristic findings on electron microscopy.
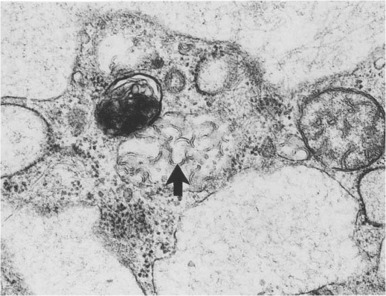
The clinical features and presence of characteristic Farber bodies on histology suggest the diagnosis of Farber disease. Definitive diagnosis relies on identification of deficient acid ceramidase activity in leukocytes or cultured skin fibroblasts or by molecular analysis.
Treatment of Farber disease is supportive (i.e., tracheostomy or resection of oral granulomas for management of upper airway obstruction). HSCT in several individuals who had classic Farber disease with neurologic involvement resulted in improved visceral features, but neurologic deterioration continued, leading to death. Several Farber disease patients without neurologic involvement have received HSCT and displayed improvement in visceral disease. These children were reported as alive and well without any evidence of neurologic deterioration. Thus individuals without neurologic involvement may benefit from HSCT.
GM 1 Gangliosidosis
GM 1 gangliosidoses are heterogeneous, progressive neurovisceral disorders resulting from a deficiency of the enzyme acid β-galactosidase because of mutations in GLB1 . There are several lysosomal β -galactosidases with different substrate specificities. In the GM 1 gangliosidoses, acid β-galatosidase deficiency leads to specific accumulation of GM 1 ganglioside, asialo-GM1 ganglioside, oligosaccharides, and keratin sulfate in neurologic and visceral tissues.
The GM 1 gangliosidoses are allelic to Morquio type B disease, a nonneurologic mucopolysaccharidosis primarily expressed as a skeletal dysplasia with a paucity of other visceral organ involvement. GM1 gangliosidosis has a phenotype similar to that of galactosialidosis, because acid β-galactosidase exists in multienzyme complex with N-acetylgalactosamine-6-sulfatase, neuraminidase, and protective protein cathepsin A (PPCA), and the defects in PPCA lead to deficiencies of all three enzymes.
GM 1 gangiosidosis presents in three forms depending on the age of onset, severity of neurologic disease, presence or absence of visceral disease, and survival: early infantile onset (type 1), late infantile onset (type 2), and adult onset (type 3). The conditions are panethnic but appear to be more common in certain populations, including those in Brazil and the Maltese Islands, and among Romani and Cypriot people. The prevalence was estimated at approximately 1 in 384,000.
Early infantile onset (type 1) GM 1 gangiosidosis, the most severe variant, usually manifests in the first 6 months after birth. In rare cases intrauterine growth retardation and oligohydramnios have been present. The most severe variant presents as fetal hydrops. More frequently, children exhibit feeding problems, failure to thrive, splenomegaly, hepatomegaly (with or without evidence of hepatic dysfunction), and coarsening of facial features. Generalized skeletal dysplasia may be present as joint stiffness. Developmental arrest occurs during the first few months, followed by progressive neurodegeneration. Cherry-red maculae are commonly observed and are an important clue to this diagnosis. Death usually occurs by 3 years of age, frequently as a result of pulmonary infections.
Late infantile or juvenile onset (type 2) GM 1 gangliosidosis manifests from approximately 6 months to 3 years of age with developmental arrest, followed by progressive neurodegeneration. The patients may exhibit other visceral organ manifestations, including hepatosplenomegaly and skeletal involvement. Cherry-red maculae are variably present. Survival may extend into the second decade.
Adult onset (type 3), or chronic, GM 1 gangliosidosis is a slowly progressive, heterogeneous CNS disease, with a variable age of onset from 3 to 30 years. Neurologic features frequently include gait disturbances, speech disturbances, dystonia, and Parkinsonism. Skeletal changes are less significant than in the more severe variants, and hepatosplenomegaly and cherry-red maculae may not be present.
Blood cell counts are generally normal. The reticuloendothelial system contains foamy histiocytes filled with strongly PAS-positive granular material. Vacuolated lymphocytes can readily be recognized in peripheral blood and bone marrow, which may contain sea-blue histiocytes. Additionally, lymphocytes and neutrophils may exhibit basophilic granules that stain mildly with toluidine blue. Tissue macrophages with grossly distended cytoplasm filled with storage lysosomes can be identified in most organs, including the liver, kidney, spleen, lymph nodes, thymus, lung, intestine, pancreas, and skeletal muscle.
GM 1 gangliosidosis is often first suspected clinically and by the detection of complex urinary oligosaccharides and glycosaminoglycans (i.e., keratin sulfate), but this is not diagnostic. Confirmation requires detection of deficient acid β-galactosidase activity in lymphocytes, fibroblasts, or plasma; however, in those with deficient acid β-galactosidase activity, subsequent neuraminidase assays are necessary to rule out galactosialidosis. Confirmation of the diagnosis may also be made by identification of a pathogenic mutation in each copy of GLB1 .
Treatment of GM 1 gangliosidosis involves management of symptoms. HSCT was unsuccessful in stabilizing or preventing neurologic deterioration in a presymptomatic child with juvenile-onset disease, despite normalization of enzyme activity in WBCs.
GM 2 Gangliosidosis, β-Hexosaminidase Deficiency Variants
The GM 2 gangliosidoses are heterogeneous neurodegenerative diseases, inherited in an autosomal recessive manner and characterized by impaired degradation of GM 2 ganglioside. This may result from deficient β-hexosaminidase activity or, more rarely, defects in the GM 2 activator protein, which is responsible for presenting the substrate to the enzyme. GM 2 ganglioside is synthesized mostly within the CNS, and consequently storage material accumulates in neuronal cells. Depending on the nature of the mutation and the biochemistry of the β-hexosaminidase enzymes, substrates can also accumulate in the visceral organs. The two isoforms of β-hexosaminidase include β-hexosaminidase A (Hex A), a heteromer consisting of α and β subunits, and β-hexosaminidase B (Hex B), a homomer of β subunits. Therefore defects in three genes, HEXA (encodes the α subunit), HEXB (encodes the β subunit), and GM2A (encodes the GM 2 activator) may result in impaired β-hexosaminidase activity. The α-chain defects lead to isolated deficiency of Hex A and the CNS disease Tay-Sachs disease. The β-chain defects lead to deficiencies of Hex A and B, because they share the common β subunit, and the CNS and visceral disease Sandhoff disease. Hex B has non-GM 2 ganglioside substrates, oligosaccharides, and glycoproteins, which occur in the viscera, and their accumulation results in hepatomegaly and skeletal defects.
The resultant phenotypes vary in age of onset, severity of neurologic disease, presence or absence of visceral disease, and age of death. Of the variants, only Hex B mutations are associated with visceral involvement. The reader is referred to other sources for further information regarding Hex A and GM2A deficiency disorders.
Sandhoff disease is panethnic with a frequency of approximately 1 in 384,000. Three variants are frequently identified, based on the age of onset, severity of disease, and survival: infantile (classic), juvenile, and adult/late onset.
Infantile onset (classic) Sandhoff disease is virtually identical to classic Tay-Sachs disease with regard to neurologic disease. Although individuals often appear normal at birth, by approximately 3 to 5 months hypotonia and exaggerated startle responses are noted. Soon afterward, a plateau in acquisition of milestones, followed by regression of development, is noted. Decreased visual attentiveness and unusual eye movements occur, and cherry-red maculae are noted on ophthalmoscopy in almost all affected individuals. After the age of 8 to 10 months, progression of the disease is rapid. Spontaneous or purposeful voluntary movements diminish, and infants becomes less responsive to their environment. Vision deteriorates rapidly, although light and dark discrimination may remain. Although not typically present at disease onset, seizures commonly develop by the age of 1 year. Progressive enlargement of the head typically begins by 18 months of age as a result of reactive cerebral gliosis and substrate deposition. In the second year decerebrate posturing, difficulties in swallowing, and increased seizures ultimately cause the affected individual to enter an unresponsive vegetative state. Death usually occurs by the age of 2 to 4 years, typically subsequent to pneumonia. Storage materials (e.g., oligosaccharides and glycosaminoglycans) accumulate in the visceral organs (i.e., spleen, liver, kidneys, bone marrow, lymph nodes) and occasionally result in organomegaly; however, evidence of visceral organ dysfunction, including hematologic abnormalities, is unusual.
Later onset disease variants (late infantile or juvenile onset and chronic or adult onset variants) present from early childhood to adulthood. The features are exceptionally variable among affected individuals but are typically milder and demonstrate slower rates of progression than those seen with the classic (early infantile onset) variant. These features may include muscle weakness, speech and swallowing problems, abnormal movements, gait disturbances, and seizures. Cherry-red maculae may be observed but are less common than in the classic (early infantile onset) variant.
On histologic examination, strongly PAS-positive storage material is observed in Kupffer cells in the liver, macrophages in the spleen, lymph nodes and lung, and renal tubular epithelium. These storage cells appear vacuolated in routine hematoxylin and eosin-stained paraffin sections. The storage material in these visceral organs is composed of heterogeneous lamellar structures, some of which resemble membranous cytoplasmic bodies.
Sandhoff disease variants are often first suspected on the basis of the clinical features and can be distinguished from other forms of GM 2 gangliosidosis by the presence of visceral involvement, including foam cells in bone marrow. Identification of deficient total (A and B) β-hexosaminidase activity and presence of a higher than normal percentage (>70%) of the Hex-S (α-chain trimers) are strongly suggestive of the diagnosis. Confirmation is made by identification of pathogenic mutations in both copies of HEXB .
Currently, the treatment for hexosaminidase B deficiency is palliative. HSCT has not reversed or stabilized neurologic involvement. Miglustat treatment of affected individuals has not forestalled neurologic deterioration.
The Mucopolysaccharidoses
The mucopolysaccharidoses are a group of inherited disorders that result from deficiency of one or more of the lysosomal enzymes required for glycosaminoglycan (GAG) or mucopolysaccharide catabolism. GAGs are long-chain complex carbohydrates that are usually linked to protein and are major constituents of connective tissue, such as cartilage and bone. Deficiencies in the specific lysosomal hydrolases needed for breakdown of chondroitin 4-sulfate, chondroitin 6-sulfate, heparan sulfate, dermatan sulfate, keratan sulfate, and hyaluronic acid result in the accumulation of incompletely degraded GAGs within the lysosome of specific cells, resulting in alterations in cell morphology and function.
Mucopolysaccharidosis Type I (Hurler Syndrome and Attenuated Mucopolysaccharidosis I)
Mucopolysaccharidosis type I (MPS I) has been divided classically into three phenotypic categories, all of which are due to deficiency in α- l -iduronidase, a soluble M6P containing enzyme. The most severe type, MPS I H, or Hurler syndrome, manifests with clinical findings during the first year but may not be recognized until the second year. Infants with MPS I H are often large for their age and may not have obvious developmental deficits until it becomes evident that language acquisition is delayed. The coarse facial features evolve over time, and review of serial photographs can be helpful in recognizing the changing phenotype. One of the earliest symptoms may be constant noisy breathing, and it is not unusual for families to complain that their child always has a cold. Lumbar prominence, termed gibbus, may be one of the earliest specific features. Hernias are common, and some children are recognized because of the failure of the initial surgical repair. Increasing head circumference, sometimes the result of a communicating hydrocephalus, develops. Corneal opacities may be present during the first year but may be difficult to see with a handheld ophthalmoscope. The skeletal abnormalities that are collectively referred to as dysostosis multiplex include oar-shaped ribs, anterior beaking of the vertebral bodies, lumbar kyphosis, and shortening of the metacarpals and phalanges. Contractures at the hips, knees, and ankles may not be recognized until the child is standing, and it is not unusual to note limitation in shoulder movement. Evolution of symptoms during the first year is usually associated with the severe form of MPS I, which includes progressive cognitive decline. The differentiation from milder forms of MPS I is crucial because of the distinct treatment options (discussed later).
The attenuated forms of MPS I, in which cognitive development is intact, have been divided historically between Scheie syndrome (onset in late childhood or adolescence) and Hurler-Scheie syndrome (onset between ages 3 and 8 years). These attenuated forms represent a continuum of severities, and referring to both as “attenuated” MPS I has become standard. It is important to remember that from the patient’s point of view none of the MPS I forms are “mild.” Individuals with later onset of clinical manifestations may present (1) to the orthopedist with joint stiffness, contracture, clawhand deformity, or carpal tunnel syndrome; (2) to the ophthalmologist with corneal clouding and glaucoma; (3) to the cardiologist with valve dysplasia; or (4) to the otolaryngologist with hearing loss or obstructed breathing.
Initial biochemical assessment usually involves quantitation and characterization of urine GAGs, which should reveal significant elevation (often 10- to 20-fold) of total GAG, predominantly heparan and dermatan sulfates. Iduronidase can be measured in both circulating WBCs and cultured skin fibroblasts. A pseudodeficiency state has been described, which can confuse the assessment of phenotypically normal family members and interfere with prenatal diagnosis using enzyme assay. Sequencing of IDUA is available and provides important information for counseling of family members as well as supporting information for the prediction of severity. Prenatal diagnosis is possible and should involve molecular as well as enzyme analysis. Since early diagnosis and treatment have improved the prognosis, there is interest in population based newborn screening. Pilot programs for the detection of MPS I and other treatable LSDs are beginning in several locations.
Treatment.
Cross-correction of lysosomal storage by application of the appropriate enzyme was demonstrated in vitro by Neufeld and coworkers. This property has been translated into treatment for MPS I using either cell-based (HSCT) or ERT therapies. HSCT from an unaffected donor has been possible for more than 20 years. Modern marrow-donor programs and transplant techniques are improving access to transplant and are fostering durable engraftment for a much larger fraction of the eligible patients. A combined approach, using ERT during the early phase of diagnosis and preparation for HSCT, is practical, safe, and without adverse effects on graft survival. Neurocognitive status is expected to decline for a few months after HSCT and then stabilize. For this reason earlier diagnosis and expedited referral for HSCT are important in maximizing eventual neurocognitive outcome. The long-term benefits for children transplanted before 2 years of age include softening of coarse features, improvement in airway manifestations, and resolution of hepatosplenomegaly. However, skeletal dysplasia persists and may progress. Short stature and the need for multiple orthopedic procedures are anticipated in transplanted individuals. Odontoid dysplasia may improve after HSCT. A role for additional ERT in well-engrafted individuals has not been defined.
Laronidase, recombinant α- l -iduronidase, was FDA approved in 2003 for the non-CNS manifestations of MPS I. Treated individuals, who require intravenous infusions weekly or biweekly, exhibit improvement in hepatosplenomegaly, joint range of motion, exercise tolerance, and biochemical markers (e.g., GAG excretion). Intrathecal enzyme therapy has been used to a limited extent for spinal complications. A trial of intrathecal enzyme during the peritransplant period is ongoing. Although intravenous ERT may have a palliative role for individuals with MPS I H, who are not candidates for HSCT, early HSCT is superior.
Gene therapy efforts using adeno-associated virus (AAV) vectors transducing human cDNA for IDUA have shown restoration of enzyme activity in brain, but no human gene therapy trials have been reported.
Small molecule approaches to restoration of endogenous activity have shown that GAG storage can be reduced in mice carrying a W392X mutation on treatment with NB84, an aminoglycoside designed to suppress translation termination at in-frame premature termination codons. In human fibroblasts in vitro, chloramphenicol, a peptidyl transferase inhibitor, has been shown to increase α- l -iduronidase activity by 100-fold, but the mechanism appears to be different from stop codon readthrough. There are currently no human trials of either of these approaches.
Mucopolysaccharidosis Type II (Hunter Syndrome)
Hunter syndrome is an X-linked recessive disorder resulting from a deficiency of iduronate-2-sulfatase. Boys with the severe variant may resemble children with the more severe variants of MPS I, although they typically present somewhat later. Many boys with Hunter disease are tall during infancy with coarse facial features, progressive evolution of developmental delay and eventual regression of milestones, stiff joints, clawhand deformity, hearing loss, and hepatosplenomegaly. Corneal clouding is absent. Behavioral difficulties, including aggression and disrupted sleep patterns, can be challenging. Diarrhea is a frequent complaint and can be difficult to manage. A specific skin feature is “peau d’orange,” a white pebbly lesion often located on the back or upper arms. Boys with the “milder” form, who do not have cognitive involvement, may present with similar somatic features and progressively develop joint stiffness, carpal tunnel syndrome, and cardiac valve disease. The less severe form is compatible with survival into adulthood and with normal cognitive achievement. However, it is by no means clinically “mild.”
The diagnosis may be first suspected on the basis of family history, clinical features, or abnormal radiologic findings. Quantitation and characterization of urine GAG excretion may be similar to findings in MPS I, and biochemical features do not predict clinical severity. Iduronate-2-sulfatase is present in plasma but can also be assayed in WBCs and cultured skin cells. Molecular confirmation is readily available, and hundreds of mutations of all types have been described. When clinical and biochemical evidence supports a diagnosis but sequencing fails to reveal a mutation, a deletion, which can involve contiguous genes, should be sought. In this case, an alternate diagnosis, such as multiple sulfatase (discussed later) deficiency, should be considered. Hunter syndrome is X-linked, and knowledge of the molecular lesion is important for genetic counseling in females at risk. Prenatal diagnosis is possible, and molecular analysis should be used as an adjunct to assays.
Treatment.
HSCT has been disappointing in MPS II, although the reason for the difference in cognitive response to HSCT (as compared to MPS I) is unclear. ERT with idursulfase was approved in 2006 to treat the non-CNS manifestations of MPS II, based on clinical endpoints of improved 6-minute walk and improved respiratory status. The therapy involves weekly intravenous infusions and requires a significant family commitment to therapy. Regular infusions have been transferred to the home setting after demonstration of safety during the first several months. Idursulfase does not enter the CNS, and ERT is not expected to have a significant impact on cognitive decline in severely affected boys. A clinical trial using the combination of intravenous and intrathecal enzyme is in progress. Gene therapy using AAV vectors has been studied in MPS II mice, with encouraging clearance of GAG is tissues and correction of skeletal abnormalities. No human gene therapy trials are in progress.
Mucopolysaccharidosis Type III (Sanfilippo Syndrome)
MPS III has predominantly CNS manifestations and can be very difficult to diagnose during the toddler years. Before age 5 the mildly coarse features can be subtle, and the striking physical findings that make MPS I and II recognizable are absent ( Fig. 25-9 ). There are four different enzymes implicated in this clinical phenotype, all of which lead to storage and urinary excretion of excessive heparan sulfate. In addition, glycosphingolipids accumulate in MPS III CNS. Phenotypic features include delayed language acquisition, hyperactivity, aggressive behavior, sleep disorders, and hirsutism. Loss of established milestones becomes evident at approximately 4 to 6 years. In general, individuals with MPS III A exhibit more rapid decline, and individuals with MPS III B have a less rapid course. The frequency of MPS III is approximately 1 in 100,000 in Western Europe, based on incidence of known cases, but many authorities suspect that MPS III may be significantly underdiagnosed.