Fig. 1
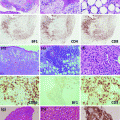
The histopathology of PTLD. a Infectious mononucleosis-like “early” PTLD lesion. b Monomorphic PTLD, Burkitt lymphoma type. c Polymorphic PTLD. d Positive EBV in situ hybridization stain in a PTLD
2.
Polymorphic PTLD (Fig. 1c). In this type, the lymphoid tissue architecture is effaced or a destructive extra-nodal mass is observed. As the name implies, the lymphoid cells are polymorphic and include small- to medium-sized lymphocytes with variable nuclear atypia, immunoblasts, and mature plasma cells. Necrosis may be observed [11]. These can be monoclonal or polyclonal, as establishing clonality is dependent on the analysis technique and clonal burden. Importantly, these do not meet diagnostic criteria for specific WHO-defined B-cell or T- /NK-cell lymphoma categories [56].
3.
Monomorphic PTLD (Fig. 1b). This is the most common type of PTLD. These are monoclonal proliferations that can be separated into specific B-cell and T-cell lymphomas, using the same WHO criteria/classification as in non-transplant patients. B-cell PTLD are more common and include diffuse large B-cell lymphoma (DLBCL), Burkitt lymphoma (BL) (Fig. 1b), plasma cell myeloma, and plasmacytoma-like PTLD. DLBCL accounts for the majority of monomorphic PTLD cases [11].
T-cell PTLD are rare and include peripheral T-cell lymphoma, hepatosplenic T-cell lymphoma, and anaplastic large cell lymphoma (ALCL). Up to 90 % of T-cell PTLD are EBV negative [57], while the majority of NK cell PTLD are EBV positive [58]. Compared to B-cell PTLD, T-cell PTLD usually occur later and carry a poorer prognosis. T-cell PTLD cases have been associated with human T-lymphotrophic virus type 1 (HTLV-1) and may be a factor in the rising incidence of T-cell lymphomas in Japan [59].
4.
Classical Hodgkin’s Lymphoma (cHL)—This is the rarest form of PTLD and usually presents late after transplantation. It is histopathologically identical to cHL, showing Reed–Sternberg cells in a variable inflammatory cell milieu.
Table 1
WHO classification of post-transplant lymphoproliferative disorders
Early lesions (5 %) | Polymorphic PTLD (15–20 %) | Monomorphic PTLD | Classical Hodgkin’s lymphoma (<5 %) | |
|---|---|---|---|---|
B-cell subtypes (>70 %) | T-cell subtypes (<5 %) | |||
Plasmacytic hyperplasia | Diffuse large B-cell lymphoma | Peripheral T-cell lymphoma, not otherwise specified | ||
Infectious mononucleosis-like | Burkitt lymphoma | Other | ||
Plasmacytoma-like | ||||
Other | ||||
An excisional tissue biopsy is the preferred specimen for PTLD diagnosis. If an excisional biopsy is not feasible, for example, in case of suspected extra-nodal involvement, core needle biopsy and needle aspiration may be diagnostic [28]. Tissue should be examined for histology, immunophenotyping, EBER-ISH (EBV-encoded small nuclear RNA—in situ hybridization), and cytogenetic studies for classification.
Basic laboratory tests should include complete blood count with differential, comprehensive metabolic panel, lactate dehydrogenase, and uric acid. Patients should also be checked for EBV viral load, HIV, and hepatitis serologies. There is insufficient data at this time to recommend following serial EBV viral loads to assess response to therapy. Modern imaging, usually computed tomography (CT) scanning with or without positron emission tomography (PET) scanning, is an essential tool in diagnosis and staging and should include neck, chest, abdomen, and pelvis. If cytopenias are present, bone marrow biopsy may be warranted [11].
Similar to Hodgkin’s and non-Hodgkin’s lymphoma in the non-transplant setting, the Ann Arbor classification is typically used for staging.
7 Prognosis
Due to the large variability of disorders encompassed in PTLD, no reliable prognostic scoring system exists. In a French study of 500 patients with PTLD post-renal transplant, the authors constructed a 5-point prognostic score based on the following: age older than 55 years, serum creatinine greater than 1.5 g/dl, elevated LDH, disseminated PTLD, and monomorphic histology [60]. Patients were risk stratified into low risk (0 risk factors), moderate risk (1 risk factor), high risk (2–3 risk factors), or very high risk (4–5 risk factors). Five-year OS was 92, 83, 59, and 25 %, respectively. Another study of 80 PTLD patients after SOT noted 3 prognostic factors: CNS involvement, bone marrow involvement, and hypoalbuminemia [61]. Three-year survival was 93 % with 0 risk factors, 68 % with 1 risk factor, and 11 % with 2–3 risk factors. CNS involvement has been associated with poor prognosis in more than one study, though this may be improving with the use of rituximab and high-dose methotrexate [11, 61].
8 Treatment
There are no uniformly applicable guidelines for the treatment of PTLD, due to the wide spectrum of disease and scarcity of prospective phase II and III studies. The goals of treatment in PTLD are twofold: first, to eliminate the PTLD and second, to preserve the transplanted graft [56]. The majority of evidence available for the treatment of PTLD has been seen in SOT (particularly CD20-positive B-cell PTLD), with limited data available regarding PTLD after allo-HCT or T-cell PTLD.
In general, the initial therapeutic intervention for PTLD consists of immune suppression reduction (ISR) [62, 63]. Unfortunately, only about half of patients respond to ISR. In addition, it can take several weeks before a response is evident after ISR [13, 64]. Many other treatment options have been studied and are used either following, or in conjunction with ISR (see Table 2 and Fig. 2). These include the use of rituximab (a monoclonal antibody directed against CD20), chemotherapy regimens, local therapy with radiation or surgery, EBV-specific CTL infusions, and more recent novel therapies.

Table 2
Phase II/III studies of PTLD treatment and outcomes
Authors | Year | N | Treatment | ORR, % (95 % CI, if available) | Median OS (mo) | |
|---|---|---|---|---|---|---|
Initial therapy (prior to study enrollment) | Subsequent (protocol) therapy | |||||
Oertel et al. [116] | 2005 | 17 | ISR (100 %) | Rituximab × 4 cycles | 59 (36–78) | 37 |
Blaes et al. [117] | 2005 | 11 | ISR + CHOP (100 %) | Rituximab × 4 cycles, repeated every 6 months until progression | 64 (35–85) | 14 |
Choquet et al. [74] | 2006 | 43 | ISR (100 %) | Rituximab × 4 cycles | 44 (30–59) | 15 |
Gonzales-Barca et al. [118] | 2007 | 38 | ISR (100 %) | Rituximab × 4–8 cycles | 66 (50–79) | 42 |
Haque et al. [97] | 2007 | 33 | ISR (100 %) + other | Allogenic EBV-specific CTL | 64 (47–78) | Not reached |
Swinnen et al. [119] | 2008 | 16 | ISR + Acyclovir (100 %) | If no CR, IFN-α daily × 3 cycles (28 days) | N/A | 19 |
If no CR after IFN, ProMACE-CytaBOM × 6 cycles | ||||||
Gross et al. [120] | 2012 | 55 | ISR (100 %) | Rituximab with CP | 69 (57–84) | Not reached |
Trappe et al. [81] | 2012 | 74 | ISR (100 %) | Rituximab × 4 cycles | 90 % | 79 |
With CR, Rituximab × 4 cycles (40 %) | ||||||
Without CR, R-CHOP × 4 cycles (60 %) |
Fig. 2
Treatment algorithm for PTLD
8.1 Immune Suppression Reduction (ISR)
In most cases of PTLD, ISR is the first step in treatment. ISR should partially restore the ability of CTLs to eliminate EBV-infected lymphocytes [8]. There are, however, several potential drawbacks associated with ISR—the most significant being graft rejection. In addition, ISR as monotherapy has a relatively slow time to response (on average>2–4 weeks) [65] and lower efficacy compared to ISR combined with rituximab and/or chemotherapy. Reported overall response rates (ORR) to ISR vary from 0 to 74 % [64, 66–68], with durable responses of less than 30 % of patients in several studies [13, 64, 66, 67].
In older studies, patients with early lesions and polyclonal PTLD appeared to respond better to ISR, while those with monoclonal tumors [46], in particular with bcl–6 expression, were less likely to respond [69, 70]. However, in a more recent study, monomorphic versus polymorphic histology did not predict for response to ISR [66].
In theory, EBV-positive PTLD should have a higher likelihood to respond to ISR; however, recent evidence suggests this may not necessarily be the case. In a recent study of SOT patients, EBV positivity was not a predictor of response—with only non-bulky disease (<7 cm) and age <50 at diagnosis being predictive [66]. Some factors such as high LDH and multi-organ dysfunction or involvement have inconsistently been associated with poor response to ISR [65, 66]. Multiple factors can be combined to help predict response to ISR. For example, using LDH elevation, hepatitis C infection, bone marrow or liver involvement, and B symptoms, 3-year overall survival (OS) was 100 % in patients with none of these factors, 79 % with one, and 8 % with two or more factors [66].
The specific dose modifications for ISR in an individual patient are based on multiple factors, such as the transplanted organ, extent of PTLD, perceived risk of rejection, symptoms, physical exam, and laboratory data. Decisions regarding ISR should be pursued in close collaboration with the transplant team. Anti-proliferative agents such as azathioprine and mycophenolate should be dose-reduced or discontinued, if possible [11]. Guidelines suggest dose reduction of 25 % in limited disease. With extensive disease in non-critically ill patients, cyclosporine/tacrolimus is typically reduced to 50 %, with discontinuation of azathioprine/mycophenolate and continuation of prednisone at 7.5–10 mg daily [71]. In critically ill patients, it may be necessary to discontinue all immune suppressing agents except prednisone 7.5–10 mg daily. Patients need to be followed closely to assess disease response and to monitor for graft rejection. Though there are risks with ISR, the majority of evidence supports the use of ISR as the first step in management of PTLD in most cases. In patients with aggressive disease, low predicted response to ISR alone or a contraindication to ISR may necessitate consideration of alternative or augmented therapies.
8.2 Rituximab
Rituximab, a chimeric monoclonal antibody targeting the B-cell surface protein CD20, has improved outcomes in many B-cell lymphomas. Data showing rituximab to be superior to other treatment approaches started to emerge around 2005 [72]. Prior to the rituximab era, the 3-year OS for PTLD ranged from 30 to 50 % but has now improved to >60 % [62, 72–75].
In the first trial of rituximab monotherapy use after ISR failure in PTLD, an ORR of 44 % was seen, with a median OS of 15 months [74]. Since then, several other studies have shown ORR ranging from 44 to 68 % and median OS up to 42 months (Table 2).
More recently, in a large, multicenter retrospective analysis, Evens et al. reviewed 80 SOT PTLD patients to establish the impact of the introduction of rituximab on outcomes [61]. All patients in the study had ISR, with 74 % of patients receiving rituximab with or without chemotherapy. With regard to first-line therapy, patients had a 73 % OS when rituximab was a part of the regimen compared to 33 % without rituximab. The 3-year progression-free survival (PFS) was 70 % with rituximab and 21 % without.
8.3 Chemotherapy
Prior to rituximab, chemotherapy was the primary treatment in patients who failed ISR. Several regimens have been described for use in PTLD. Anthracycline-based chemotherapy with rituximab, such as R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) is the most widely used regimen [71, 76–78]. It is preferred in patients for whom a rapid clinical response is required, such as those with multi-organ involvement or rapidly declining clinical status. Although the risk of toxicity and treatment-related mortality (TRM) can be as high as 25–50 % [79], the ORR is higher than rituximab monotherapy, ranging between 65 and 100 % [71]. Given the increased risk of TRM, chemotherapy should be reserved for those who fail to achieve appropriate response with ISR or ISR and rituximab, who have highly aggressive histology, or who require a rapid clinical response such as patients presenting with severe organ dysfunction or rapidly declining clinical status [71]. In some scenarios, it may be prudent to avoid certain chemotherapy drugs if possible, such as anthracyclines in heart transplant patients and high-dose methotrexate in renal transplant patients.
8.4 Sequential Therapy
Many experts recommend a sequential or staged approach to the treatment of PTLD, in which patients generally receive ISR or ISR + rituximab as a starting point. An assessment of response is made 4–8 weeks later and, if the response is insufficient, additional therapy is then administered [63, 71, 80, 81]. This strategy was recently tested in a large prospective study (the “PTLD-1” trial) and may provide the foundation for a standard approach in the future [81]. In this study, 70 patients who failed ISR were then treated with 4 weekly doses of rituximab followed by 4 cycles of CHOP. Following rituximab, there was a 60 % ORR (with 20 % CR). After completion of rituximab × 4 and CHOP × 4, the ORR increased to 90 % (with 68 % CR) [81]. In this study, the planned therapy was rituximab × 4 followed by CHOP × 4, so it is unclear whether the patients who achieved CR with rituximab alone might have maintained their responses without chemotherapy.
In practice, a commonly utilized approach is to initially implement ISR or ISR + rituximab and then, for those who have not achieved CR, to subsequently apply chemotherapy. The “sequential therapy” approach of the PTLD-1 trial was modified to test this “risk-adapted” approach, after an interim analysis demonstrated that response to rituximab correlated with OS [82]. In the “risk-adapted” approach, patients achieving CR with 4 weeks of rituximab received 4 additional doses of rituximab, whereas those who did not achieve CR were then treated with 4 cycles of R-CHOP. In a preliminary analysis (n = 90), an ORR of 93 % and CR rate of 78 % were seen. The CR rate to rituximab alone was 27 %, with an additional 51 % going on to achieve CR after CHOP. Only 13 % of patients achieving CR with rituximab alone went on to relapse. Treatment-related mortality was low at 8 %. Comparing to previous data from the “sequential therapy” part of the PTLD-1 trial, it appears that (1) omitting chemotherapy from those achieving CR to rituximab is safe, and (2) for those who have progressive disease with rituximab, R-CHOP is more effective than CHOP. Those who failed to achieve CR after rituximab × 4 ± R-CHOP × 4 (22 %) did not benefit from additional R-CHOP and had nearly 80 % risk of progressive lymphoma in the following 1–2 years. Therefore, with this approach, most patients were able to achieve CR, while the relatively high treatment-related toxicity seen with CHOP was avoided except for patients who truly required chemotherapy [82]. Given these promising results, further study may help establish this “response-adapted” approach as a standard of care in CD20-positive PTLD [63].
8.5 Localized Therapy
Localized therapy using either radiation therapy (RT) or surgery can be utilized in carefully selected PTLD patients. When combined with ISR, this may be curative in patients with PTLD localized to a single site (Ann Arbor Stage I disease) [63]. RT may also have a significant role in the treatment of CNS or limited-stage PTLD, with some studies demonstrating complete responses [83, 84]. Incorporation of RT may also allow for an abbreviated course of chemotherapy for those with limited-stage disease or avoidance of chemotherapy for those who are unlikely to tolerate chemotherapy.
8.6 Primary CNS PTLD
Primary CNS PTLD is unique in both its presentation and treatment compared to other forms of PTLD. CNS involvement occurs in up to 15–22 % of PTLD, most of which are primary CNS PTLD, and has consistently been associated with poor outcomes across multiple studies [6, 12, 61, 75, 84–87].
In a recent multicenter international retrospective analysis of 84 cases of primary CNS PTLD over a 14-year period, it was found that although 83 % of cases occurred late (>1 year post-transplant), over 90 % were EBV positive [87]. The large majority (79 %) were associated with renal transplantation, presented with a median time to onset of 54 months, and 79 % had diffuse large B-cell lymphoma (DLBCL) histology. Median PFS and OS for the cohort were 8 months and 17 months, respectively.
There are no clear guidelines for the treatment of primary CNS PTLD. Options include the use of ISR, rituximab, high-dose methotrexate (HD-MTX), high-dose cytarabine, and whole brain radiation [63]. The role of rituximab is poorly defined in this setting, given its poor penetration across the blood brain barrier [71]. Response to first-line therapy is the most important prognostic factor [85, 87]. In a review of 289 patients with primary CNS PTLD after SOT, it was reported that 32 of the 39 cases of CR occurred in patients who received RT [84]. Overall, however, the outcomes were poor, with <20 % of patients achieving long-term remission. In general, treatment with ISR alone is rarely effective for primary CNS PTLD. For now, treatment should be approached in a manner akin to the primary CNS lymphoma, including whole brain RT, HD-MTX, and high-dose cytarabine, bearing in mind that HD-MTX is likely to have increased toxicity in renal transplant patients [71, 87, 88].
8.7 Burkitt PTLD
Burkitt PTLD accounts for less than 5 % of PTLD cases [63]. It is a highly aggressive malignancy with frequent extra-nodal manifestations, high proliferative index, high latent EBV infection, and rapid tumor growth. Translocations involving c–myc and the heavy or light chain immunoglobulin loci underlie the aggressive biology. Burkitt Lymphoma, in the non-transplant population, has been successfully treated with short-duration intensive combination chemotherapy, along with aggressive CNS prophylaxis [89].
Unfortunately, there are no consensus guidelines or treatment protocols for the treatment of Burkitt PTLD. Due to the aggressive nature of the disease, ISR alone or ISR + rituximab are not recommended. Instead, most clinicians proceed directly to a combined approach involving ISR, rituximab, and chemotherapy. In a small study, CR was achieved in 5/5 patients prospectively treated with ISR and R-CHOP [90]. The role of intrathecal chemoprophylaxis is unclear in Burkitt PTLD. However, extrapolating from the non-transplant setting, we recommend CNS prophylaxis in cases of Burkitt PTLD as well. Highly aggressive regimens typically used for BL in the non-transplant setting should be used with extreme caution in PTLD patients due to the increased risk for toxicity [91].
8.8 Hodgkin’s and Hodgkin-like PTLD
Hodgkin PTLD is the rarest form of PTLD; as a result, treatment data are limited to a few case reports and series [92, 93]. The approach to treatment for Hodgkin PTLD is similar to that employed in Hodgkin’s lymphoma (HL). This typically involves chemotherapy or combined modality treatment using chemotherapy and RT [94]. The most commonly used chemotherapy regimen for HL in the USA is ABVD (doxorubicin, bleomycin, vinblastine, dacarbazine). In one study of 7 patients with Hodgkin PTLD treated with standard HL therapy along with ISR, 4 patients achieved CR [93]. In another series, 4 out of 4 patients treated with standard regimens achieved CR [92].
8.9 Anti-viral Therapy
Anti-viral agents, such as acyclovir and ganciclovir, have been studied as either prophylaxis or treatment for PTLD. These agents require activation by EBV thymidine kinase (EBV-TK), an enzyme expressed in infected replicating cells, and are therefore ineffective against latent EBV-infected B cells [95, 96]. Arginine butyrate can be used to induce EBV-TK in infected latent B cells, with subsequent administration of ganciclovir to eliminate these cells. Results utilizing arginine butyrate and ganciclovir are encouraging [95], but further trials are required to prove clear clinical benefit. Prophylactic use of these agents has had mixed results [71]. Outside of clinical trials, anti-viral agents are not currently recommended for routine prophylaxis or treatment of PTLD.
Related posts:
 Hematopoietic Cell Transplantation in Non-Hodgkin’s Lymphomas
Hematopoietic Cell Transplantation in Non-Hodgkin’s Lymphomas
 and Therapy of Chronic Lymphocytic Leukemia and Small Lymphocytic Lymphoma
and Therapy of Chronic Lymphocytic Leukemia and Small Lymphocytic Lymphoma
 of Non-Hodgkin Lymphomas: Diagnosis and Response-Adapted Strategies
of Non-Hodgkin Lymphomas: Diagnosis and Response-Adapted Strategies
 Expression Profiling in Non-Hodgkin Lymphomas
Expression Profiling in Non-Hodgkin Lymphomas
 Therapeutic Strategies and New Treatment Paradigms for Follicular Lymphoma
Therapeutic Strategies and New Treatment Paradigms for Follicular Lymphoma
 of T-Cell Lymphomas: Diagnosis and Biomarker Discovery
of T-Cell Lymphomas: Diagnosis and Biomarker Discovery
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


