Lymphomas
Urban Novak
Laura Pasqualucci
Riccardo Dalla-Favera
The term lymphoma identifies a heterogeneous group of biologically and clinically distinct neoplasms that originate from the lymphoid organs and have historically been divided into two distinct categories, namely non-Hodgkin’s lymphoma (NHL) and Hodgkin’s lymphoma (HL).1,2 During the past 3 decades, significant progress has been made in elucidating the molecular pathogenesis of lymphoid malignancies as a clonal malignant expansion of B cells (in the majority of cases) or of T cells. The molecular characterization of the most frequent genetic abnormalities associated with lymphoma development has led to the identification of a number of protooncogenes and tumor suppressor genes that are altered in B-cell NHL (B-NHL) and whose abnormal functioning contributes to lymphoma pathogenesis. Relatively less is known about the pathogenesis of T-cell NHL (T-NHL) and HL. This chapter will focus on the molecular pathogenesis of the most common types of lymphoma, including B-NHL, T-NHL, HL, and chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), which also derives from mature B cells. Emphasis will be given to the mechanisms of genetic lesion and the nature of the involved genes in relationship to the normal biology of lymphocytes.
THE CELL OF ORIGIN OF LYMPHOMA
Lymphomas originate from mature B cells in approximately 85% of the cases, while the remaining 15% derive from the T-cell lineage. A key concept for the understanding of lymphomagenesis is the relationship between these tumors and the unique DNA modification events that take place in normal lymphocytes in order to enable the production of highly efficient neutralizing antibodies in B cells, and to encode T-cell receptors in T cells.
Normal B-Cell Development and the Dynamics of the Germinal Center Reaction
B lymphocytes are generated from a common pluripotent stem cell in the bone marrow, where precursor B cells first assemble their immunoglobulin heavy chain locus (IGH) followed by the light chain loci (IGL) through a site-specific process of cleavage and rejoining, known as V(D)J recombination.3,4 Cells that fail to express a functional (and nonautoreactive) antigen receptor are eliminated within the bone marrow, while B-cell precursors that have successfully rearranged their antibody genes are positively selected to migrate into peripheral lymphoid organs as mature, naive B cells.5 In most B cells, the subsequent maturation steps are linked to the histologic structure of the germinal center (GC), a highly specialized microenvironment that forms following encounter of naive B cells with a foreign antigen, together with signals from CD4+ T and antigen-presenting cells.5,6,7
The development of the GC can be schematically described as occurring in two stages. First, B cells enter the GC dark zone, which consists of rapidly proliferating centroblasts (CBs) (doubling time <12 hours). In this phase, CBs modify the variable region of their Ig genes (IgV) by the process of somatic hypermutation (SHM), which introduces mostly single nucleotide substitutions but also deletions and duplications in order to change their affinity for the antigen.5,7,8,9,10 CBs express elevated levels of BCL6,11,12 a transcriptional repressor13 that negatively regulates a broad set of genes, including those involved in (1) B-cell receptor (BCR) and CD40 signaling14 (2) T-cell mediated B-cell activation14; (3) induction of apoptosis14,16; (4) response to DNA damage, by modulation of genes involved in the sensing and execution of DNA damage responses17,18,19,20; (5) multiple cytokine and chemokine signaling pathways, including the ones involved in interferon and transforming growth factor-β responses14,16; and (6) plasma cell differentiation, via suppression of the PRDM1/BLIMP1 master regulator.21,22,23,24 This transcriptional program suggests that the BCL6 function is critical to establish the proliferative status of CBs while allowing the execution of DNA modification processes (SHM and class-switch recombination) without eliciting responses to DNA damage, and preventing premature activation and differentiation prior to the selection for the survival of cells producing high affinity antibodies.
In the light zone, CBs are thought to cease proliferation and differentiate into centrocytes (CCs), which are rechallenged by the antigen through the
interaction with CD4+ T cells and follicular dendritic cells.5,6 CCs expressing a BCR with reduced affinity for the antigen are eliminated by apoptosis, while a few cells with high affinity will be stimulated by a variety of signals, including the engagement of their BCR by the antigen itself and the activation of the CD40 receptor by the CD40 ligand present on CD4+ T cells. These signals down-regulate BCL6, allowing the arrest of proliferation and the restoration of DNA damage responses, as well as activation and differentiation capabilities, such that B cells can be selected for survival and differentiation into memory cells and plasma cells.6,25 In the GC, CCs also undergo class-switch recombination (CSR), a DNA remodeling event that confers distinct effector functions to the antibodies.26 Both SHM and CSR depend on the activity of the activation-induced cytidine deaminase (AID) enzyme and represent B-cell-specific functions that modify the genome of B cells via mechanisms involving single- or double-strand breaks,27,28,29 a notion that will become important in the understanding of the mechanisms generating genetic alterations in B-NHL.
interaction with CD4+ T cells and follicular dendritic cells.5,6 CCs expressing a BCR with reduced affinity for the antigen are eliminated by apoptosis, while a few cells with high affinity will be stimulated by a variety of signals, including the engagement of their BCR by the antigen itself and the activation of the CD40 receptor by the CD40 ligand present on CD4+ T cells. These signals down-regulate BCL6, allowing the arrest of proliferation and the restoration of DNA damage responses, as well as activation and differentiation capabilities, such that B cells can be selected for survival and differentiation into memory cells and plasma cells.6,25 In the GC, CCs also undergo class-switch recombination (CSR), a DNA remodeling event that confers distinct effector functions to the antibodies.26 Both SHM and CSR depend on the activity of the activation-induced cytidine deaminase (AID) enzyme and represent B-cell-specific functions that modify the genome of B cells via mechanisms involving single- or double-strand breaks,27,28,29 a notion that will become important in the understanding of the mechanisms generating genetic alterations in B-NHL.
This schematic description is useful to focus on two key concepts for the understanding of B-NHL pathogenesis. First, the activity of SHM, which introduces irreversible DNA changes in the genome, allowed to conclude that most B-NHL types, with the exception of mantle-cell lymphoma (MCL), derive from GC-experienced B cells, as they contain hypermutated IgV sequences, and that clonal expansion occurred within the GC, because the malignant clones contain largely identical mutations, suggesting the derivation from a single founder cell.30 Second, the most frequent oncogenic events in B-NHL—namely, chromosomal translocations and aberrant somatic hypermutation (ASHM)—result from mistakes in the machinery that normally diversifies the Ig genes during B lymphocyte differentiation, further supporting the GC origin of most B-NHL (Fig. 30.1). Finally, the definition of two distinct phases during GC development reflects stages of B-cell differentiation and function that can to some extent be recognized in different B-NHL subtypes.
Normal T-Cell Development
T-cell development proceeds through sequential stages defined according to the expression of the molecules CD4 and CD8. Committed lymphoid progenitors exit the bone marrow and migrate to the thymus as early T-cell progenitors or double-negative 1 (DN1) cells, which lack expression of CD4 and CD8 as well as of the T-cell receptor (TCR).31 In the thymic cortex, T cells advance through the double-negative stages DN2, DN3, and DN4, while undergoing specific rearrangements at the TCRβ locus in order to acquire expression of the pre-TCR.31 Those thymocytes that have successfully recombined the pre-TCR will be selected to further differentiate into doublepositive cells (DP; CD4+CD8+), which express a complete surface TCR and can then enter a process of positive and negative selection in the medulla, before exiting the thymus as single positive T cells.31 The end result of this process is a pool of mature T cells that exhibit coordinated TCR and coreceptor specificities as required for effective immune responses to foreign antigens. Most mature T-NHLs arise from postthymic T cells in the lymphoid organs.
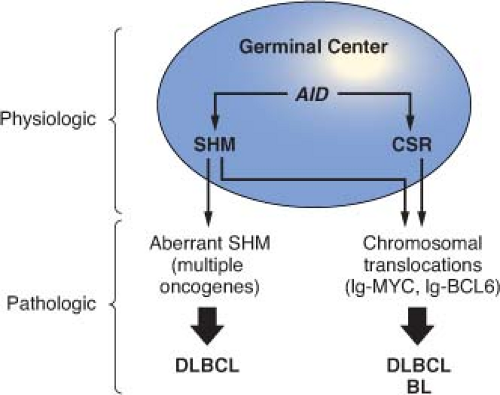 FIGURE 30.1 Model for the generation of genetic lesions during lymphomagenesis. B-NHL-associated genetic lesions appear to be due to mistakes occurring during the physiologic processes of somatic hypermutation and class-switch recombination in the highly proliferative environment of the germinal center (top). These include chromosomal translocations, which in most cases juxtapose the Ig genes to one of several protooncogenes (e.g., BCL6 or MYC), and aberrant somatic hypermutation of multiple target genes AID, activation-induced cytidine deaminase; SHM, somatic hypermutation; CSR, class-switch recombination. |
GENERAL MECHANISMS OF GENETIC LESION IN LYMPHOMA
Chromosomal Translocations
Chromosomal translocations are the genetic hallmark of malignancies derived from the hematopoietic system. Lymphoma-associated translocations represent reciprocal and balanced recombination events that occur between two specific chromosomes, are clonally represented in each tumor case, and are often recurrently associated with a given tumor type.
Although the precise molecular mechanisms that are responsible for the generation of translocations
remain partially obscure, significant advances have been obtained during the past decade in our understanding of the events that are required for their initiation.32 It has now been documented that chromosomal translocations occur at least in part as a consequence of mistakes during Ig and TCR gene rearrangements in B and T cells, respectively. Based on the characteristics of the chromosomal breakpoint, three distinct scenarios can be distinguished: (1) translocations derived from mistakes of the RAG-mediated V(D)J recombination process, as is the case for translocations involving IGH and CCND1 in MCL or IGH and BCL2 in follicular lymphoma (FL)32,33,34; (2) translocations mediated by errors in the AID-dependent CSR process, such as those involving the Ig genes and MYC in sporadic Burkitt lymphoma (BL)32; and (3) translocations occurring as by-products of the AID-mediated SHM mechanism, which also generates DNA breaks, such as those joining the Ig and MYC loci in endemic BL.32 Conclusive experimental evidence for the involvement of antibodyassociated remodeling events has been recently provided through in vivo studies performed in lymphoma-prone mouse models, where the removal of the AID enzyme was sufficient to abrogate the generation of MYC-IGH translocations in normal B cells undergoing CSR35,36 and to prevent the development of GC-derived B-NHL.37,38
remain partially obscure, significant advances have been obtained during the past decade in our understanding of the events that are required for their initiation.32 It has now been documented that chromosomal translocations occur at least in part as a consequence of mistakes during Ig and TCR gene rearrangements in B and T cells, respectively. Based on the characteristics of the chromosomal breakpoint, three distinct scenarios can be distinguished: (1) translocations derived from mistakes of the RAG-mediated V(D)J recombination process, as is the case for translocations involving IGH and CCND1 in MCL or IGH and BCL2 in follicular lymphoma (FL)32,33,34; (2) translocations mediated by errors in the AID-dependent CSR process, such as those involving the Ig genes and MYC in sporadic Burkitt lymphoma (BL)32; and (3) translocations occurring as by-products of the AID-mediated SHM mechanism, which also generates DNA breaks, such as those joining the Ig and MYC loci in endemic BL.32 Conclusive experimental evidence for the involvement of antibodyassociated remodeling events has been recently provided through in vivo studies performed in lymphoma-prone mouse models, where the removal of the AID enzyme was sufficient to abrogate the generation of MYC-IGH translocations in normal B cells undergoing CSR35,36 and to prevent the development of GC-derived B-NHL.37,38
The common feature of all NHL-associated chromosomal translocations is the presence of a protooncogene in proximity to the chromosomal recombination sites. In most lymphoma types, and in contrast with acute leukemias, the coding domain of the oncogene is not affected by the translocation, but its pattern of expression is altered as a consequence of the juxtaposition of heterologous regulatory sequences derived from the partner chromosome (protooncogene deregulation) (Fig. 30.2). Two distinct types of protooncogene deregulation (i.e., homotopic and heterotopic) can be distinguished. Homotopic deregulation occurs when the protooncogene becomes constitutively expressed in the lymphoma cell, while its expression is tightly regulated in normal lymphoid cells. Conversely, heterotopic deregulation occurs when the protooncogene is not expressed in the normal tumor counterpart and undergoes ectopic expression in the lymphoma. In most types of NHL-associated translocations, the heterologous regulatory sequences responsible for protooncogene deregulation are derived from antigen receptor loci that are expressed at high levels in the target tissue.32 However, in certain translocations, such as the ones involving BCL6 in diffuse large B-cell lymphoma (DLBCL), different promoter regions from distinct chromosomal sites can be juxtaposed to the protooncogene in individual tumor cases, a concept known as promiscuous translocations.39,40,41,42,43,44,45,46
Less commonly, B-NHL-associated chromosomal translocations juxtapose the coding regions of the two involved genes to form a chimeric unit that encodes for a novel fusion protein, an outcome typically observed in chromosomal translocation associated with acute leukemia (Fig. 30.2). Examples are the t(11;18) of mucosa-associated lymphoid tissue (MALT) lymphoma and the t(2;5) of anaplastic large cell lymphoma (ALCL). The molecular cloning of the genetic loci involved in most recurrent translocations has led to the identification of a number of protooncogenes involved in lymphomagenesis (Table 30.1).
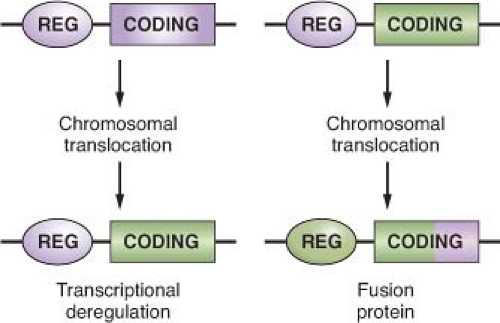 FIGURE 30.2 Molecular consequences of chromosomal translocations. Top panel: schematic representation of the two protooncogenes involved in prototypic chromosomal translocations, with their regulatory (REG) and coding sequences. Only one side of the balanced, reciprocal translocations is indicated. Bottom panel: two distinct outcomes of chromosomal translocations. In the case of transcriptional deregulation (left scheme), the normal regulatory sequences of the protooncogene are substituted with regulatory sequences derived from the partner chromosome, leading to deregulated expression of the protooncogene. In most cases of B-NHL, the heterologous regulatory regions derive from the Ig loci. In the case of fusion proteins (right scheme), the coding sequences of the two involved genes are joined in frame into a chimeric transcriptional unit that encodes for a fusion protein, characterized by novel biochemical and functional properties. |
Aberrant Somatic Hypermutation
The term aberrant somatic hypermutation (ASHM) defines a recently identified mechanism of genetic lesion that is uniquely associated with B-NHL, particularly DLBCL, leading to the mutation of multiple non-Ig genes.47 ASHM has been proposed to derive from a malfunction in the physiologic SHM process, although the mechanism involved in this malfunction has not been identified.
In GC B cells, SHM is tightly regulated both spatially and temporally to introduce mutations only in the rearranged IgV genes8 as well as in the 5′ region of a few other genes, including BCL6 and the CD79 components of the B-cell receptor,48,49,50,51,52 although the functional role of the mutations found in these other genes remains obscure. On the contrary, multiple mutational events were found to affect numerous loci in over 50% of DLBCL cases, as well as in a few other lymphoma types, including, among others, AIDS-associated B-NHL, primary central nervous system lymphomas, and posttransplant lymphoproliferative disorders.53,54,55,56,57 The identified target loci comprise more than 10% of the genes transcribed in B cells and include several well-known protooncogenes such as PIM1 and MYC, one of the most frequently altered human oncogenes.47 These mutations are typically distributed within ˜2 Kb from the transcription initiation site (i.e., the hypermutable domain in the Ig locus)58 and, depending on the genomic configuration of the target gene, may affect nontranslated as well as coding regions, thus altering the response to factors that normally regulate their expression, or changing key structural and functional properties.47 This is the case of MYC, where a significant number of events lead to amino acid changes with proven functional consequences in activating its oncogenic potential. However, a comprehensive characterization of the potentially extensive genetic damage caused by ASHM is still lacking.
TABLE 30.1 MOST COMMON GENETIC LESIONS ASSOCIATED WITH NON-HODGKIN’S LYMPHOMA (NHL) | ||||
|---|---|---|---|---|
|




