Lymphoid Tissues and Organs
Eitan M. Akirav
Noelia Alonso-Gonzalez
Lucy A. Truman
Nancy H. Ruddle
INTRODUCTION
The mammalian immune system defends against invading pathogens, by both the innate and adaptive mechanisms. Although cells that can respond to pathogens are scattered in tissues throughout the body, the optimal structures for the response to antigens are organized, compartmentalized cellular aggregates that facilitate antigen concentration and presentation to a large repertoire of antigen-specific lymphocytes. The primary lymphoid organs, the fetal liver, thymus, and bone marrow, are the sites where diverse populations of naïve lymphocytes mature to disperse throughout the body to await foreign invaders. This remarkable differentiation process occurs in a foreign antigen-independent fashion. The secondary lymphoid organs, including the lymph nodes, spleen, Peyer’s patches, and other mucosal-associated lymphoid tissues (MALTs), are discrete sites in which naïve, antigen-specific T- and B-lymphocytes encounter invaders to generate an adaptive response. The lymph nodes and spleen have been considered to be somewhat static structures, while, in fact, they are responsive to environmental influences and undergo remarkable changes in the course of antigenic challenge. Precise programs control the development of the spleen, lymph nodes, Peyer’s patches, tonsils and adenoids, and (in the mouse and rat) the nasal-associated lymphoid tissue (NALT). Somewhat less anatomically restricted tissues that are even more sensitive to the environment facilitate and include accumulations of lymphoid cells are organized, but less discretely defined: the bronchus-associated lymphoid tissues (BALTs) and inducible lymphoid follicles (ILFs). Tertiary lymphoid organs, or more accurately, tertiary lymphoid tissues, are accumulations of lymphoid cells that arise ectopically in sites that are not anatomically restricted and are not regulated by developmental programs. Tertiary lymphoid tissues respond to environmental stimuli and arise during chronic inflammation subsequent to microbial infection, graft rejection, autoimmunity, or cancer by the process of lymphoid neogenesis.1,2 In this chapter, the structure, function, trafficking patterns, and developmental signals that regulate the hierarchy of lymphoid organs will be described.
PRIMARY LYMPHOID ORGANS
The primary lymphoid organs are the sites where pre-B- and pre-T-lymphocytes mature into naïve B and T cells in the absence of foreign antigen. Each T cell or B cell expresses a unique receptor that can recognize and respond to exogenous antigen and, in most cases, discriminate between self-and foreign antigens. Naïve cells leave the primary lymphoid organs having received and responded to developmental cues that result in the rearrangement of their genetic material to generate a repertoire capable of recognizing and responding to a wide variety of foreign antigens. In the course of maturation in the primary lymphoid organs, the naïve lymphocytes express various chemokine receptors and adhesion molecules that direct them to secondary lymphoid organs.
Fetal Liver
The earliest lymphoid cell precursors derive from self-renewing hematopoietic precursors called hematopoietic stem cells (HSCs). During ontogeny, these cells occupy several niches. In the fetal mouse, the first wave of hematopoiesis occurs in the yolk sac and aorta-gonad-mesonephros region at E10.5.3 The placenta also contains HSC activity.4,5,6 Cells leave these tissues and migrate to the fetal liver, and then the bone marrow and thymus and spleen under the influence of chemokines and adhesion molecules (Fig. 3.1). The cells in the fetal liver respond to CXCL12 (stromal cell derived factor) and, in contrast to those in the bone marrow, also respond to Steel factor7 (Table 3.1). The fetal liver is also the source of CD4+CD3- lymphoid tissue inducer cells that express lymphotoxin (LT)α (also called tumor necrosis factor [TNF] β) and LTβ. The requirement of inducer cells for the
development of secondary lymphoid organs is described in the following. Differentiation of HSCs to B cells occurs in the fetal liver, but does not require interleukin (IL)-7. B-cell development is described in detail elsewhere in this volume. Of the several different subsets of B cells, those generated by the fetal liver HSCs are somewhat limited and are of the B-1 subset.8 They do not give rise to cluster of differentiation (CD)5 B cells, predominately B1-B cells, and do not express terminal deoxynucleotidyl transferase and myosin-like light chain.8,9 It is not known whether these differences are intrinsic to the cells or are due to differences in the cytokine environment of fetal liver and bone marrow. The ligands for P-selectin, E-selectin, and vascular cell adhesion molecule (VCAM)-1 are required for the cells to leave the fetal liver and home to the bone marrow.10,11
development of secondary lymphoid organs is described in the following. Differentiation of HSCs to B cells occurs in the fetal liver, but does not require interleukin (IL)-7. B-cell development is described in detail elsewhere in this volume. Of the several different subsets of B cells, those generated by the fetal liver HSCs are somewhat limited and are of the B-1 subset.8 They do not give rise to cluster of differentiation (CD)5 B cells, predominately B1-B cells, and do not express terminal deoxynucleotidyl transferase and myosin-like light chain.8,9 It is not known whether these differences are intrinsic to the cells or are due to differences in the cytokine environment of fetal liver and bone marrow. The ligands for P-selectin, E-selectin, and vascular cell adhesion molecule (VCAM)-1 are required for the cells to leave the fetal liver and home to the bone marrow.10,11
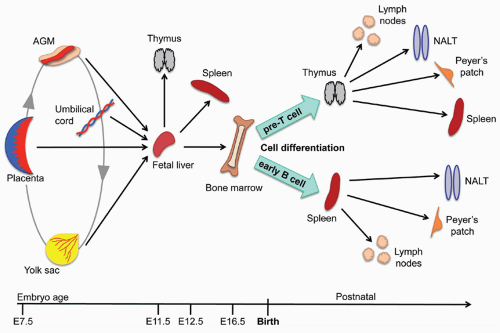 FIG. 3.1. Embryonic and Postnatal Development of T and B Cells in Mouse Primary and Secondary Lymphoid Organs. Embryonic diagram based on information summarized in Medvinsky et al.6 |
TABLE 3.1 Chemokines Implicated in Lymphoid Organ Development and Maintenance | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||
Bone Marrow
Functions
The bone marrow is the source of self-renewing populations of stem cells. These cells include hematopoietic precurscor cells, HSCs, and endothelial progenitor cells, which may derive from a single precursor.12 The adult bone marrow contains stem cells that can differentiate into adipocytes, chondrocytes, osteocytes, and myoblasts.13 Collectively, these cells are called hematopoietic/stem progenitor cells. In the adult, cells leave the leave the bone marrow and seed the thymus where they undergo differentiation to naïve T cells. Additional factors enable the differentiation of immature B cells from HSCs.
In addition to serving as a primary lymphoid organ where B-cell differentiation and development occur, the bone marrow is also a home for antibody secreting cells.14 After B cells
have interacted with antigen in the secondary lymphoid organs, such as the lymph nodes, spleen, and Peyer’s patches, they enter the bloodstream and travel to the marrow. Thus, this organ not only serves as a primary lymphoid organ, but also as a reservoir for fully differentiated plasma cells.
have interacted with antigen in the secondary lymphoid organs, such as the lymph nodes, spleen, and Peyer’s patches, they enter the bloodstream and travel to the marrow. Thus, this organ not only serves as a primary lymphoid organ, but also as a reservoir for fully differentiated plasma cells.
Architecture: Cellular and Functional Niches
The microenvironment of the bone marrow, contained in the central cavity of bone, is a complex three-dimensional structure, with cellular niches that influence B cells during their development and later, as plasma cells, as they return to the bone marrow. The bone marrow has a rich blood supply with a nutrient artery that branches into ascending and descending arteries further dividing into cortical capillaries, periosteal capillaries, and endosteal capillaries, finally merging into a sinus.15 Previously, the prevailing understanding of B-cell differentiation in the bone marrow was that primitive HSCs were located in close contact with the endosteum near osteoblasts (the “endosteal niche”). During the course of differentiation into mature B cells, they moved into the central region of the bone marrow cavity (the “vascular niche”).16 The former niche was identified as the location of HSC; the latter, as the site of B-cell differentiation.17 This anatomic concept has been challenged, as it has been reported that HSCs are found throughout the bone marrow. More recently, a reticular niche has been described that includes CXCL12 abundant reticular cells.18 Growth factors and cytokines produced by different stromal cells influence cells at different stages in their differentiation. Thus, it is more appropriate to consider functional or cellular, rather than anatomical, niches.15,19 Once the HSCs differentiate into immature B cells expressing cell surface immunoglobulin (Ig) M, they undergo processes of negative selection and receptor editing, leave the bone marrow, and travel through the blood stream to the secondary lymphoid organs where they complete their differentiation.
Several cytokines and chemokines influence B-cell differentiation in the bone marrow. Flt-3 ligand (also called Flk-2L) signals B-cell differentiation and growth and synergizes with several other hematopoietic growth factors.20 Its receptor, Flt-3 (also known as Flk-2), expressed by primitive HSCs, is a member of the class II tyrosine kinase family. In contrast to fetal liver, HSCs from adult bone marrow do not respond to Steel factor. Chemokines contribute to B-cell differentiation in the bone marrow and define the functional niches. For excellent reviews of this topic, see Nagasawa,15 Heissig et al.,18 and Mazo et al.21 Many of these factors affect other lymphoid cells, such as dendritic cells (DCs) and T cells. Several of these factors, whose functions have been identified in gene deletion studies in mice, in morphologic analysis, and in cell culture studies, play roles in multiple aspects of lymphoid organ development; their activities, though important in the bone marrow, are not limited to that organ. CXCL12, also known as stromal cell derived factor, is a chemokine that is crucial for recruitment of HSCs to the bone marrow. It is widely expressed by osteoblasts, reticular cells,19 and endothelial cells.22 In fact, the interaction of HSCs expressing CXCR4, the receptor for CXCL12, with that chemokine on the endothelial surface is the first step in the HSC’s exit from the circulation into the marrow.23 CXCL12 is also essential for the earliest stage of B-cell development (pre-pro-B cells). Its receptor, CXCR4, is expressed on early B cells and is downregulated in pre-B cells. CXCR4 remains at low levels in immature B cells and mature B cells in secondary lymphoid organs, but is upregulated after B cells interact with antigen and differentiate into plasma cells.14 This explains the propensity of antibody-secreting cells to return to the bone marrow. Once a pre-pro-B cell has interacted with CXCL12, it moves on to a different cell expressing IL-7. In B-cell development, IL-7 acts later than CXCL12 in a narrow window between pro-B cells and immature B cells in the scheme proposed by Nagasawa.15
Traffic In and Out: Chemokines and Adhesion Molecules
The extensive vascularization of the bone marrow allows entrance of hematopoietic precursors and plasma cells and egress of mature cells. Hematopoietic progenitor recruitment to the bone marrow in the mouse is dependent on the interaction of a variety of chemokines, integrins, and selectins, and their receptors, counter receptors, and vascular cell adhesion molecules. These include α4 integrin (VLA4 or α4β1), VCAM-1,24 P-selectin glycoprotein ligand-1, E-selectin,25 α4β7, and mucosal addressin cell adhesion molecule (MAdCAM-1).26 A small subpopulation of newly formed B cells in the bone marrow that expresses L-selectin has been described,27 suggesting a mechanism for entrance into lymph nodes (see subsequent discussion).
In addition to the acquisition of Ig expression that occurs in the bone marrow under the control of stromal cells, B cells express various chemokine receptors and adhesion molecules in the process of differentiation. In a study of human bone marrow B cells, it was determined that pro-B and pre-B cells migrate toward CXCL12, but not toward a wide range of other chemokines, including CCL19 and CCL21, though they do express low levels of CCR7, the receptor for those ligands. However, mature bone marrow B cells do respond to CCL 19 and CCL 21,28 CXCL13 (indicating a functional CXCR5), and CCL20 (MIP3α). On the other hand, CCR6 expression and responsiveness to CCL20 are only seen in mouse B cells after they have migrated into the periphery during the process of maturation and are in the circulating B cell pool.29
Thymus
Functions
The thymus is defined as a primary lymphoid organ due to its inimitable role in T-cell development. Indeed, a mutant known as the “nude” (nu/nu) mouse, which lacks a normal thymus, is completely devoid of mature T cells.30 Although some studies suggest that T cells can develop extrathymically in organs such as the “cervical thymus”31 and the gut epithelium,32 the thymus remains the main site for T-cell maturation, education, and selection. T-cell precursors represent more than 95% of total cells in the thymus and give rise to mature T cells. These cells are crucial components of the adaptive immune system in that they are highly specific in their ability to recognize a nearly infinite number of antigens owing to their diverse repertoire.30,33
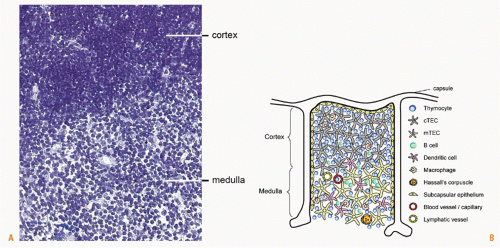 FIG. 3.2. A: Thymic structure. Low power magnification of a hematoxylin-stained frozen section of a mouse thymus. The cortex is well populated with lymphocytes; the medulla is less tightly packed and thus stains less intensely. B: Diagram of mouse thymic cellular populations. Precursor T cells enter through blood vessels at the corticomedullary junction. They progress to the medulla where they undergo differentiation from double negative to double positive (DP) cells expressing T-cell receptors. Thymic stromal cells provide growth factors. DP cells undergo positive selection, under the influence of cortical thymic epithelial cells. Single positive (SP) cluster of differentiation (CD)4 or CD8 cells migrate into the medulla where they undergo negative selection, mainly through autoimmune regulator expressing medullary thymic epithelial cells and dendritic cells. SP cells, having undergone differentiation, exit through blood vessels (not shown) to the periphery. |
The T-cell repertoire is shaped during development in the thymus by the processes of positive and negative selection. Negative and positive selection are so stringent that nearly 95% of all T-cell precursors are deleted in the thymus.34 Negative selection ensures that T cells, which are capable of recognizing the organism’s self-antigens presented on major histocompatibility complex (MHC) class I or II molecules with high affinity, are eliminated prior to their export to the periphery. In contrast, positive selection, which requires a higher degree of T-cell receptor (TCR) avidity than negative selection,35 allows for T cells recognizing self-antigen with low to medium affinity to leave the thymus and eventually protect the organism against invading pathogens.
While negative selection is highly efficient in eliminating the majority of self-reactive T cells, some of these cells do escape the thymus and exit to the periphery. These autoreactive cells impose a threat to various organs as their activation may result in the development of autoimmune diseases. The immune system has evolved several ways to prevent the activation of autoreactive T cells in the periphery. One mechanism that protects against autoimmunity is the generation of a T-cell population capable of suppressing activation of self-reactive T cells. These thymus-derived protective T cells, known as regulatory cells (Treg), recognize self-antigens with relatively high affinity.
Architecture
The thymus in the mouse consists of two symmetric lobes located above the heart, while in humans the thymus is multilobed. Each lobe can be divided into three distinct regions: capsule, cortex, and medulla. The latter two regions harbor thymocytes at various maturation stages. Although maturing T cells constitute the majority of cells in the thymus, other cell types such as macrophages, DCs, B cells, and epithelial cells are also present.36 Histologic analysis of the thymus reveals a clear distinction between the thymic cortex and medulla, which are separated by a corticomedullary border. The thymic cortex appears darker and more densely populated with T-cell precursors, whereas, the medulla appears considerably lighter and contains smaller numbers of T cells relative to other cell types (Fig. 3.2A). Blood vessels and small blood capillaries are found throughout the thymus. The fact that T-cell progenitors are found in the more highly vascularized corticomedullary border suggests that blood vessels in this region facilitate the entry of T-cell progenitors into the thymic parenchyma.37 In the thymic medulla, the close association between medullary thymic epithelial cells and thymic blood vessels38 suggests that these vessels may act as organizers of the medullary thymic compartment. Lymphatic vessel distribution coincides with that of blood vessels and capillaries.39 The majority of lymphatic vessels are located in the thymic medulla, though some can also be found in the cortex. The role of lymphatic vessels in thymic function is unclear, although it has been proposed that these vessels deliver extrathymic antigens into the thymus or export mature T cells from the thymus into the circulation.
Cellular Composition and Functions
T-cell precursors at various stages of differentiation represent the majority of cells in the thymus. Antigen-presenting cells (APCs) of either hematopoietic or stromal origin mediate the education and selection of T cells in the thymus. The different stages in T-cell selection and maturation that take place in distinct regions of the thymus (Fig. 3.2B) are discussed at length elsewhere in this volume. Briefly, lymphoid progenitors enter the thymus at the corticomedullary border.37 Following their entry, these cells, identified as double negative (DN) T cells due to a lack of expression of the cell surface molecules CD4 and CD8, undergo four maturation steps termed DN1 to DN4, which are distinguished by the expression of two additional cell surface molecules: CD25 and CD44.40,41 DN3 cells migrate to the subcapsular zone while rearranging their TCRβ chain and expressing it in combination with a surrogate α chain. Those cells that have successfully rearranged the genes for α and β chains of the TCR become double positive (DP) cells, and express both CD4 and CD8 surface markers. In the cortex, DP cells undergo negative and positive selection.42,43 Positively selected DP cells further differentiate into single positive (SP) cells expressing either CD4 or CD8. Following their differentiation, SP cells relocate to the medulla where they mature and undergo further rounds of deletion. SP cells that do survive are then exported out of the thymus.44 DN precursors can give rise to an additional T-cell population expressing the γδTCR. These T cells are distinct from TCRαβ T cells in their tissue distribution and recognition of antigens. TCRαβ DP cells control the development of TCRγδ cells via the production of LTβ.45 Treg development in the human thymus occurs at the DP stage allowing for the production of CD4 and CD8 positive Tregs.46
The thymic parenchyma consists of a complex threedimensional structure supported by thymic epithelial cells (TECs). TECs in the thymic cortex and thymic medulla are phenotypically and functionally distinct and support different stages of T-cell maturation. Several cell surface markers are used to distinguish medullary TECs (mTECs) from cortical TECs (cTECs) in the mouse. Among these markers are the cytokeratins K5 and K8, the adhesion molecule Ep-CAM, and the glycoprotein Ly-51. K8 and Ly-51 are expressed by cTECs, while K5 and Ep-CAM are expressed by mTECs.47,48 TECs are unique in that they express MHC II constitutively, similarly to professional APCs. The role of TECs in T-cell selection was recently elucidated by their expression of the transcription factor, autoimmune regulator (AIRE). This transcription factor plays an important role in T-cell selection and prevention of autoimmunity, as illustrated by the fact that humans with a mutated form of the AIRE gene exhibit polyendocrine autoimmunity due to inadequate T-cell selection.49,50 It has become clear that AIRE controls the expression of certain tissue-restricted antigens in the thymus, such as insulin, a protein that is unique to the β cells of the islets of Langerhans in the pancreas.48,51 The expression of tissue-restricted antigens in the thymus facilitates the negative selection of maturing T cells, which would otherwise be allowed to leave the thymus. The thymic medulla includes distinct structures also known as Hassall corpuscles. Originally described by Arthur Hill Hassall in 1849, this structure consists of concentric stratified keratinizing epithelium. Hassall corpuscles are implicated in several processes of the thymus,52 including the expression of tissue-restricted antigens, such as insulin (a key antigen in type 1 diabetes),53 Igs, and filaggrin (key antigens in rheumatoid arthritis),54,55 and serving as a prominent site for T-cell apoptosis.56
DCs are professional APCs that are found in the thymus. Thymic DCs can be divided into two distinct populations. The first population originates from a thymocyte precursor,57,58 whereas the second population is derived from partially mature peripheral DCs that continuously enter the thymus from the circulation.59 DCs do not appear to be involved in positive selection, but do contribute to negative selection.60,61,62,63 They have also been implicated in the selection of Tregs in humans,64 and recent reports in the mouse suggest that peripheral DCs can migrate to the thymus and serve as efficient inducers of Tregs.65 Activation of thymic DCs can be mediated in part by the IL-7-like cytokine, thymic stromal lymphopoietin. In the human thymus, Hassall corpuscles produce thymic stromal lymphopoietin, and thymic stromal lymphopoietin-activated DCs can alter the fate of self-reactive T cells from deletion to positive selection of Tregs.66 More specifically, thymic stromal lymphopoietin expression in a subset of DCs, plasmacytoid DCs, can induce the generation of particularly potent Tregs from CD4+CD8- CD25- thymocytes.67 These findings highlight the heterogeneity of DCs and emphasize their ability to fulfill different roles during T-cell development in the thymus.
Macrophages and B cells are additional hematopoieticderived professional APCs in the thymus. In contrast to DCs, thymic macrophages are located throughout the thymus and do not play a significant role in T-cell selection.68 B cells are detected in human and mouse thymus at relatively low numbers69,70 and are characterized by the expression of the cell surface molecule CD5. They are capable of producing antibodies of several different isotypes.71 It has been suggested that thymic B cells induce negative selection in developing thymocytes,69 although B cell-deficient mice show a limited T-cell repertoire when compared with normal mice, also suggesting a role in positive selection.72 Recent data consistent with a role for B cells in thymic negative selection have emerged. Mice whose B cells were experimentally manipulated to express a myelin oligodendrocyte glycoprotein peptide show deletion of myelin oligodendrocyte glycoprotein-specific T effector cells.73 One mechanism for the effect of B cells on T-cell selection is suggested by the observation that B cells play a role in the control of tissue-restricted antigens. Studies done in mice deficient in B cells showed fewer thymic epithelial cells with reduced expression of insulin and myelin oligodendrocyte glycoprotein, native antigens of the pancreas and brain, respectively.74
Traffic In and Out: Adhesion Molecules and Chemokines
Adhesion molecules mediate the extravasation of leukocytes from blood and lymphatic vessels into the thymus. These molecules also play an important role in facilitating lymphocyte homing into various regions of the thymus. Indeed,
thymic blood vessels express the adhesion molecules intercellular adhesion molecule (ICAM)-1, VCAM-1, CD34, peripheral node addressin (PNAd) and vascular adhesion protein-1.75 The expression of high levels of ICAM-1, VCAM-1, and vascular adhesion protein-1 on venules near the corticomedullary border suggests that these molecules may play a role in the recruitment of thymocyte progenitors. More specifically, vascular adhesion protein-1, which is restricted to the venules surrounding the sites of progenitor homing, can mediate the extravasation of leukocytes. The regional distribution of these adhesion molecules further illustrates the importance of a distinct anatomical separation between cortex and medulla and represents their individual functions.
thymic blood vessels express the adhesion molecules intercellular adhesion molecule (ICAM)-1, VCAM-1, CD34, peripheral node addressin (PNAd) and vascular adhesion protein-1.75 The expression of high levels of ICAM-1, VCAM-1, and vascular adhesion protein-1 on venules near the corticomedullary border suggests that these molecules may play a role in the recruitment of thymocyte progenitors. More specifically, vascular adhesion protein-1, which is restricted to the venules surrounding the sites of progenitor homing, can mediate the extravasation of leukocytes. The regional distribution of these adhesion molecules further illustrates the importance of a distinct anatomical separation between cortex and medulla and represents their individual functions.
Chemokine-mediated T-cell migration and traffic to and within the thymus is crucial for normal T-cell selection. The compartmentalization of the thymus is orchestrated by a milieu of chemokines. The thymic cortex is involved in the maturation of DN cells to DP cells and by positively selecting DP T cells capable of recognizing MHC:peptide complexes and eliminating the cells that are not. The thymic medulla acts as a site of negative selection of SP T cells based on TCR recognition avidity to self-antigens and possibly positive selection of Treg. Various chemokines that are produced by the thymic cortex and medulla allow T cells expressing different chemokine receptors to home to specific regions of the thymus. This differential expression of chemokines is complemented by the fact that T cells at different maturation states express different chemokine receptors. During development, the entry of lymphoid progenitors into the thymus is highly dependent on CCL21 and CCL25, which bind the chemokine receptors CCR7 and CCR9, respectively. Mice lacking CCR7 or CCL2176,77 show a transient delay in thymus colonization by lymphocytes (day 14.5), and this delay is further extended (day 17.5) in mice lacking CCR9.78 Lymphoid progenitors enter the thymus at the corticomedullary border and commence their migration outwards toward the subcapsular region of the cortex as DN3 cells. The expression of the chemokine receptors CXCR4 and CCR7 by DN cells is important in directing cell migration.79,80 In the subcapsular region, DN thymocytes that have successfully rearranged their TCRαβ chains progress to the DP cell stage. Positively selected DP cells move inwards toward the thymic medulla for further differentiation into SP cells. The ligands for CCR7 are crucial in mediating the migration of positively selected DP cells into the medulla as illustrated by the fact that a deficiency in CCR7 or its ligands, CCL19 or CCL21, prevents DP cell relocation from the cortex to the medulla resulting in abnormal central tolerance.81,82 This abnormal tolerance is associated with a reduction in thymic B-cell numbers and reduced expression of tissue-restricted antigens.74 Interestingly, antigen itself can control T-cell migration speed from the cortex to the medulla. In the presence of a negative selection ligand, T cells slow down considerably and are limited to a confined zone 30 µ in diameter allowing for a more prolonged selection and induction of developmental arrest.83
The export of positively selected SP T cells out of the thymus is also dependent on chemokines. Chemokines involved in T-cell emigration are CXCL12 and its receptor CXCR4, which repel SP cells out of thymus,84 and CCL19, which promotes T-cell emigration from the thymus of newborn mice.85 The chemoattractant, sphingosine 1-phosphate (S1P), is an additional mediator of T-cell emigration. SP T cell express a S1P receptor (S1P1) and are attracted to the high levels of S1P present in the serum promoting their egress.86,87
While the role of different thymic compartments and chemokines in the maturation of naïve SP T cells has been extensively studied, the thymic regions and chemokines that control the selection of regulatory T cells remain largely unknown. It may be that chemokines produced by both cTECs and thymic DCs play a role in regulatory T-cell selection, albeit during different stages of maturation.
Development
The initial development of the thymus at midgestation in the mouse is independent of vascularization or bone marrow-derived cells. In the mouse, the thymus rudiment is first evident on day 11 of gestation as it evolves from the endoderm of the third pharyngeal pouch.88 This gives rise to the thymic lobes as well as to the parathyroid gland. On day 12.5 of gestation, a separation of the primordium is observed and by day 13.5 a distinct thymus is apparent. Evidence has suggested that not only the pharyngeal pouch endoderm but also the ectoderm may also be involved in thymic development.89,90 More specifically, it was suggested that the pharyngeal pouch ectoderm contributes to the development of cTECs whereas the endoderm contributes solely to the development of mTECs. This “dual origin” model was mainly supported by histological data as well as data collected from thymi of nu/nu mice.91,92,93 More recently, it was shown that both thymic cortex and medulla are derived solely from the pharyngeal pouch endoderm94; although the endoderm and ectoderm are found in close proximity between gestational day 10.5 and 11, only the endoderm actively contributes to thymic development.95 These findings support the so-called single origin model of thymus development.
The contribution of lymphocytes to the normal development of thymic cortex and medulla is well recognized. In models of T-cell deficiency, cTEC and mTEC development is halted at different stages depending on the stage of T-cell arrest. If T-cell development is arrested at the DN stage, as in the recombination activating gene-deficient mouse, the thymic medulla is greatly reduced while the thymic cortex remains unaffected.96,97 A more severe phenotype is observed in transgenic mice that overexpress the human CD3 signaling molecule. In these mice, T-cell arrest occurs earlier than in recombination activating gene knockout mice, leading to a loss of both cortex and medulla and to a shift from a three-dimensional to a two-dimensional structure of the thymic epithelia.97
Recently, LTα and LTβ have been identified as master regulators of mTEC development and expression of tissue-restricted antigens. LTα and LTβ are members of the TNF superfamily and mediate the processes of secondary lymphoid organ development and inflammation.1,98 In the absence of LT, tissue-restricted antigen and AIRE expression are reduced and certain mTEC subpopulations fail to develop.99,100 T cells, owing to their vast numbers, appear
to be the main source for LT production in the thymus45; however, additional resident cells also serve as a source of LT. Recently, resident thymic B cells were identified as the highest source of LTα and LTβ on a per cell basis,74 revealing yet another aspect of thymic B cells in thymus development and function.
to be the main source for LT production in the thymus45; however, additional resident cells also serve as a source of LT. Recently, resident thymic B cells were identified as the highest source of LTα and LTβ on a per cell basis,74 revealing yet another aspect of thymic B cells in thymus development and function.
The development of the thymus in humans closely follows the model of thymic development in the mouse and bird. Similar to the mouse, human thymic colonization by hematopoietic stem cells occurs relatively early, at week 8.2 of gestation. During this stage, the thymic medulla and cortex are organized, suggesting that thymocytes are required for normal thymic development. Between gestation week 9.5 and 10, the first signs of thymocyte negative selection are evident, and by gestation week 10 to 12.75 the gradual onset of positive selection is detected.101
SECONDARY LYMPHOID ORGANS
Naïve cells express their receptors for specific antigen, leave primary lymphoid organs, circulate through the bloodstream, migrate into the tissues, and lodge in secondary lymphoid organs. The frequency of naïve cells specific for an individual antigen is quite low (estimates range from 1 in 105 to 1 in 106). Thus, the chance that an individual T or B cell will encounter its specific antigen in the circulation is rather low. Secondary lymphoid organs are strategically located in anatomically distinct sites where foreign antigen and APCs efficiently concentrate and activate rare antigen-specific lymphocytes, thus leading to the initiation of adaptive immune responses and generation of long-lived protective immunity. These organs include highly organized, compartmentalized, and mostly encapsulated tissues such as lymph nodes, spleen, appendix, tonsils, murine NALTs, and Peyer’s patches. Naïve cells are also primed in less discrete tissues throughout the body, including the BALTs, cryptopatches, and ILF.
Lymph Nodes
Lymph nodes are bean-shaped structures dispersed along lymphatic vessels. The lymphatic vessel system plays important roles in tissue fluid balance, fat transport, and the immune response. In contradistinction to blood vessels, which form a closed recirculating system, lymphatic vessels comprise a blind-end, unidirectional transportation system. The absorbing lymphatic vessels, or lymphatic capillaries, remove interstitial fluid and macromolecules from extracellular spaces and transport the collected lymph through the primary collector. The collected lymph and its cellular contents are transported into the thoracic duct and returned back to blood circulation. In humans, lymph collected from the entire lower body region, the left head, and left arm region accumulates in the thoracic duct and returns to blood circulation via the left subclavian vein; lymph collected from right head and right arm region returns to blood via the right subclavian vein.
Lymph nodes, usually embedded in fat, are located at vascular junctions, and are served by lymphatic vessels that bring in antigen, and connect them to other lymph nodes. Though most lymph nodes are classified as peripheral lymph nodes, a few (cervical, mesenteric, and sacral), termed mucosal nodes, express a slightly different complement of endothelial adhesion molecules, cooperate with the mucosal system, and are regulated somewhat differently in development (see following discussion). Although all lymph nodes are vascularized, and thus can receive antigens from the bloodstream, they are also served by a rich lymphatic vessel system and are thus particularly effective in mounting responses to antigens that are present in tissues. These antigens may be derived from foreign invaders that are transported by APCs or can be derived from self-antigens. Thus, lymph nodes extend the role of the primary lymphoid organs and discriminate between dangerous foreign antigens and benign self-antigens. This capacity relies on the APCs and their state of activation in the lymph node, and the recognition capacity of the naïve T and B cells.
Lymph nodes can also function as niches for generating peripheral tolerance, an additional mechanism to minimize the effects of those self-reactive T-cells that escape central tolerance in the thymus.102 DCs constitutively sample selfantigens and migrate to draining lymph nodes even in the steady state.103,104,105 Because most self-antigen-bearing DCs in lymph nodes are immature106 and have low levels of costimulatory molecules, they are not effective at activating naïve cells. They regulate self-reactive T cells by inducing anergy, clonal deletion, and/or expanding Tregs.105,106,107,108,109 Several groups have recently described populations of cells in secondary lymphoid organs that can present tissue-restricted antigens to CD8 cells.110,111,112,113,114 These include two different populations of AIRE-expressing stromal cells, DCs, and AIRE-negative lymphatic endothelial cells. These cells may play a role in self-tolerance, or perhaps they can prime for autoimmunity in inflammatory conditions.
Structure and Organization
A collagen capsule surrounds the highly compartmentalized lymph node (Fig. 3.3). The cortical region includes discrete clusters called primary follicles consisting of densely packed naïve B cells and follicular DCs (FDCs). After B cells encounter their cognate antigen, they are activated. They then proliferate secondary follicles and germinal centers develop. T cells and DCs distribute in the paracortex. Macrophages reside in the subcapsular zone and in the medullary area, and those in the subcapsular zone appear to be particularly adept at presenting antigen-antibody complexes to B cells.115 Follicular DCs, a population of mesenchymal origin, support B-cell follicles or germinal centers under stimulation.1 Plasma cells are also concentrated in the medulla as they prepare to leave the lymph node and circulate to the bone marrow. A network composed of reticular fibers, fibrous extracellular matrix bundles, and another mesenchymal cell population, the fibroblastic reticular cells, supports the entire lymph node.116 Compartmentalization of cells in the lymph node is orchestrated by lymphoid chemokines CCL19, CCL21, and CXCL13. Stromal cells in the paracortical region produce CCL19 and the protein is transported to the surface of high endothelial venules (HEVs).117 CCL21 is encoded by several genes118; CCL21-leu is expressed by lymphatic vessels
outside the lymph node119; CCL21-ser is made by stromal cells and HEVs in the lymph node. CCL19 and CCL21 recruit CCR7-expressing cells across the HEVs to the paracortical region. CXCL13, produced by stromal cells in the B-cell follicles, attracts CXCR5 expressing B cells.120 After naïve T and B cells encounter antigen, they undergo extensive changes in expression of chemokine receptors and adhesion molecules that result in their movement to different areas of the lymph node or leaving it all together.121,122 S1P1 facilitates lymphocyte egress from lymph nodes as they move toward the ligand S1P in the lymph,123,124 as discussed in more detail subsequently.
outside the lymph node119; CCL21-ser is made by stromal cells and HEVs in the lymph node. CCL19 and CCL21 recruit CCR7-expressing cells across the HEVs to the paracortical region. CXCL13, produced by stromal cells in the B-cell follicles, attracts CXCR5 expressing B cells.120 After naïve T and B cells encounter antigen, they undergo extensive changes in expression of chemokine receptors and adhesion molecules that result in their movement to different areas of the lymph node or leaving it all together.121,122 S1P1 facilitates lymphocyte egress from lymph nodes as they move toward the ligand S1P in the lymph,123,124 as discussed in more detail subsequently.
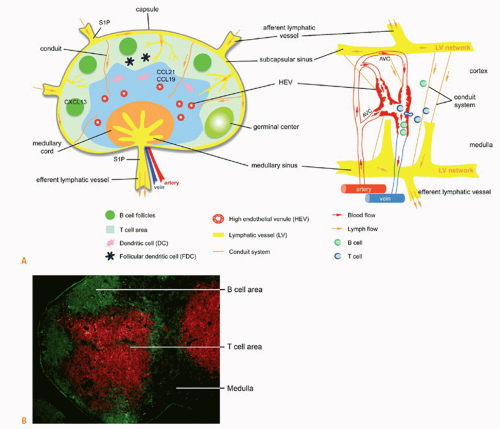 FIG. 3.3. A: Lymph node structure and functional regions. The lymph node is divided into an outer cortex and inner medulla surrounded by a capsule and lymphatic sinus. The cortex includes B cells and follicular dendritic cells. The paracortical region includes T cells and dendritic cells. Macrophages are found in the subcapsular sinus and medullary cord. Lymphocytes enter into the lymph node through an artery at the hilus region, and into the parenchyma through high endothelial venules (HEVs) expressing peripheral node addressin and CCL19 and CCL21. Stromal cells also produce these chemokines. T cells and dendritic cells (DCs) are directed to the paracortical region by CCL19 and CCL21. B cells are directed to the cortex by CXCL13. After interaction with antigen and T cells, germinal centers develop. Antigen and DCs drain into the lymph node from the tissues through afferent lymphatic vessels. Antigen continues to percolate through the node via a conduit system. Activated cells leave through efferent lymphatic vessels. Diagram of the blood network of a rat lymph node (right) is adapted from Anderson and Anderson.125 Note the arteriovenous communications, the venous sphincters, and cells leaving the HEVs into the parenchyma. B: Lymph node compartmentalization. Immunofluorescent staining of B cells (anti-B220, green) in follicles in the cortex and T cells (anti-cluster of differentiation 3, red) in the paracortical area. The medulla is unstained. |
Lymph Node Vasculature: Blood Vessels and Lymphatic Vessels
Soluble antigen and APCs enter into lymph nodes via afferent lymphatic vessels. After surveying antigens in the lymph
nodes, lymphocytes leave those organs via efferent lymphatic vessels that can connect to the next lymph node in the chain and finally return to blood circulation.125,126,127 In this manner, HEVs and lymphatic vessels maintain lymphocyte homeostasis during the steady state.
nodes, lymphocytes leave those organs via efferent lymphatic vessels that can connect to the next lymph node in the chain and finally return to blood circulation.125,126,127 In this manner, HEVs and lymphatic vessels maintain lymphocyte homeostasis during the steady state.
Blood endothelial cells play a crucial role in lymphocyte trafficking in the lymph node. One or two arteries enter the lymph node at the hilus. These arteries branch and pass through the medulla area, enter the cortex, and sometimes continue in the subcapsular area. Beneath the subcapsular sinus, the branching capillaries form loops and some of them become arteriovenous communications. Arteriovenous communications become HEVs in the cortex area and occasionally extend from the subcapsular sinus to the medulla. HEVs constitute a specialized postcapillary network in the lymph node, playing a critical role in lymphocyte recirculation. Each main HEV trunk receives three to five branches lined with high endothelial cells and two or three branches lined with flat endothelial cells. The luminal diameters of HEVs progressively increase from cortex to medulla. Finally, HEVs merge into segmental veins in the medulla area and join larger veins in the hilus125 (see Fig. 3.3A). Intravital microscopy has revealed that the entire venular tree consists of five branching orders with the higher orders in the paracortex and the lower orders located in the medulla and hilus areas. Only the higher order venules, located in the T-cell area, are specialized into HEVs and are recognized by the monoclonal antibody MECA-79.126,127 Recirculating naive lymphocytes leave the bloodstream via HEVs, specialized vessels with a high cuboidal endothelium, and migrate into the lymph node parenchyma. PNAd, defined by the MECA-79 antigen, is an L-selectin ligand, and is a characteristic HEV adhesion molecule. PNAd is composed of a variety of core glycoproteins, including GlyCAM-1, CD34, Sgp200, and podocalyxin; these proteins must be sialylated, sulfated, and fucosylated to become functional L-selectin ligands. The several enzymes that mediate these posttranslational modification events include FucT-IV, FucT-VII, and GlcNAc6ST2 (also called HEC-6ST, LSST, GST-3, HECGlcNAc6ST, gene name Chst4),128,129,130,131,132 which with the exception of a population of cells in the intestine133,134 is uniquely expressed in high endothelial cells. Together with another sulfotransferase, GlcNAC6ST1, that is expressed at sites in addition to HEVs, it sulfates glycoproteins in the Golgi apparatus135 to generate the MECA-79 epitope. PNAd, expressed on the endothelial surface, slows down (tethers) naïve L-selectinhi lymphocytes in their progress through the blood vessels. After this initial interaction, CCL19 and CCL21 on the HEVs are instrumental in activating the lymphocyte integrin lymphocyte function-associated antigen (LFA)-1. This results in tight binding of LFA-1 to ICAM-1 on the HEV, facilitating diapedesis of lymphocytes that migrate between or through the endothelial cells toward the chemokines located in the paracortical region (T cells, DCs) or cortex (B cells). Data suggest that the HEV-lymphocyte interaction is not random in different lymphoid tissues in the mouse.136,137 T-lymphocytes adhere preferentially to peripheral lymph node HEVs, B cells prefer to adhere to HEVs in Peyer’s patches, and T and B cells exhibit an intermediate pattern of adhesion to mesenteric lymph node HEVs.34,136,138 The selective adhesion of naïve lymphocytes to HEVs is at least partially controlled by the differentially expressed adhesion molecules in different lymphoid organs. PNAd rapidly replaces MAdCAM-1 after birth in mouse peripheral lymph nodes,139 but is expressed in mucosal lymph nodes together with MAdCAM-1, the ligand for the integrin α4β7.
Lymphatic vessels also play critical roles in the immune response. The collected lymph and cell contents enter the lymph node via several afferent lymphatic vessels and filter through the node where they again are concentrated in the medullary sinus. Lymphatic vessels are concentrated in the subcapsular sinus and medullary area.125 Factors from afferent lymph can be either transported deep into the lymph node cortex or move via the subcapsular sinus and leave the lymph node through efferent lymphatic vessels.116 In this manner, soluble antigen and APCs from peripheral tissues are efficiently concentrated in the draining lymph node and initiate an adaptive immune response. Lymphocytes can also enter lymph nodes through afferent lymphatic vessels and leave via efferent vessels and move to the next lymph node in the chain. This is accomplished in part through a gradient of S1P, which is at a high concentration in the lymph and low concentration in the lymph node. Lymphocytes in the lymph node downregulate this receptor (S1P1) and then upregulate it as they prepare to leave and migrate toward the higher concentration in the efferent lymph.123,124 Human and murine lymphatic vessels express several characteristic markers: lymphatic vessel endothelial hyaluronan receptor-1 (LYVE-1), Prox-1, podoplanin, CCL21, the vascular endothelial growth factor receptor-3 (VEGFR-3), and neuropilin-2.140
Conduit System
A conduit system in the lymph nodes physically connects the lymphatic sinus with the walls of blood vessels and enables the incoming factor(s) from lymph to move rapidly deep into the paracortical area.116,125 The conduit system consists of four layers: 1) a core of type I and type III collagen bundles; 2) a microfibrillar zone composed largely of fibrillins; 3) a basement membrane abundant with laminins 8 and 10, perlecan, and type IV collagen that provides a supportive structure; and 4) fibroblastic reticular cells that embrace the entire conduit system.141,142 This conduit system enables incoming lymph to penetrate deep into the T-cell area. A special subset of immature DCs, called conduit-associated DCs, can take up and process antigens moving along the conduit.142 In this manner, the conduit system probably provides a physical support for rapid initiation of adaptive immune responses after immunization.
The Intimate Relationship Between Lymph and High Endothelial Venules
Incoming lymph is necessary for the maintenance of HEV phenotype and function. After afferent lymphatic vessels are severed, dramatic changes occur in HEVs. These include flattening of the endothelium, a decrease in the uptake of 35S-sulphate143,144 (a functional marker of GlcNAC6ST-2), a reduction of lymphocyte adherence to the vessels,145,146,147
and decreased expression of PNAd and the HEV genes, Glycam-1 and Fuc-TVII.146,147,148 An increase in MAdCAM-1 expression146 suggests that these events are not simply due to a general downregulation of blood vessel gene expression and indicates that continual accumulation of afferent lymph factor(s) in lymph nodes is necessary for HEV maintenance. The nature of such lymph factor(s) is unknown.
and decreased expression of PNAd and the HEV genes, Glycam-1 and Fuc-TVII.146,147,148 An increase in MAdCAM-1 expression146 suggests that these events are not simply due to a general downregulation of blood vessel gene expression and indicates that continual accumulation of afferent lymph factor(s) in lymph nodes is necessary for HEV maintenance. The nature of such lymph factor(s) is unknown.
Topographic relations between HEVs and the lymphatic sinus have been described in the rat mesenteric lymph nodes. Some HEVs are located in the medulla area and positioned closely in relation to the lymphatic sinus.125 Occasionally, HEVs are separated from an adjacent lymphatic sinus by only a thin layer of collagen bundles. The closely apposed HEV and lymphatic sinus in the medulla provide the physical support for the intimate relationship between lymph and HEV. However, most HEVs are located in the paracortical area and are separated from the lymphatic sinus by lymphocytes. The conduit system physically connects the subcapsular sinus and HEVs and allows incoming lymph factor(s) to migrate rapidly to the wall of HEVs.125,149,150 Low-molecular-weight fluorescent tracers (below 70 kD) move rapidly via the conduit system and lead directly to the wall of HEVs. In this manner, low-molecular-weight tracers can migrate with lymph within minutes to HEVs and enter the HEV lumen.116 Lymph-borne chemokines likely adopt this route to regulate HEV function. IL-8 administration via afferent lymph increases lymphocyte HEV transmigration within minutes.125,151 In addition, lymph-borne chemokines, such as MIP-1α, also enter the conduit system and move rapidly to HEVs.116 These data suggest that lymph factor(s) can quickly access and regulate HEVs.
Development
Despite their distinctions in the adult with regard to morphology and expressed genes, a close association of blood vessels and lymphatic vessels is seen during embryogenesis. The generation of embryonic lymphatic vessels from preexisting veins in pig embryos was first described in the early 1900s by Sabin and has recently been molecularly defined.140 A variety of transcription factors have been identified that contribute to the lymphatic specification and maintenance of the lymphatic vessels phenotype.152 As early as mouse E9, expression of Sox18 and CoupTFII is apparent in the cardinal vein. These induce Prox1 in the dorsolateral side of the vein resulting in polarization and lymphatic-biased endothelial cells. Prox1 generates a feedback signal for the further maintenance of budding and migration of endothelial cells. Targets of Prox1 include Prox1 itself, podoplanin, VEGFR3, integrin-α, and Nrp-2. Prox1 expression in blood endothelial cells also represses expression of markers of those cells. NfatC1, Foxc2, and Tbx1 are additional transcription factors that are expressed later in lymphatic vessel development and contribute to patterning, pericyte covering of lymphatic vessels, lymphatic vessel valves, and lymphatic vessel maturation. At E11.5-12.0, CCL21 is expressed in these lymphatic-biased endothelial cells, as is VEGFR-3, which is reduced in blood endothelial cells. The endothelial cells expressing LYVE-1, Prox1, VEGFR-3, and CCL21 become irreversibly committed toward a lymphatic pathway.153,154 The separation of lymphatic endothelial cells from venous endothelium requires a Syk/SLP-76 signal155 that is provided by platelets.156,157 Thus, during early lymphangiogenesis, some endothelial cells express both blood vessel and lymphatic vessel markers, indicating the close association of these two vascular systems. Mesenchymal lymphangioblasts may also contribute to early lymphangiogenesis.158 The lymphatic venous junction remains in adults in only limited regions but plays an essential role in connecting the function of the two vascular systems.
Studies in the mouse have taken advantage of transgenic, knockout, and imaging studies to provide a mechanistic understanding of the process of lymph node development. Although lymph sacs per se do not appear to be essential for the initiation of lymph nodes, a lymphatic vessel system is necessary for the later development and cellular population of lymph nodes.159 Furthermore, an interaction between mesenchymal cells and primitive endothelial cells is apparent in the early lymph node anlage.160
Cytokines, Chemokines, and Transcription Factors in Lymphoid Organogenesis
The TNF-/LT-receptor family members play key roles in secondary lymphoid organ development. LTα3, signaling through TNFRI and TNFRII, and membrane bound LTα1β2, signaling through the LTβ receptor (LTβR) have been implicated. Mice deficient in LTα lack all lymph nodes and Peyer’s patches, and exhibit a disorganized spleen and severely disorganized NALT (see subsequent discussion).161,162,163 Mice deficient in LTβ lack peripheral lymph nodes but retain mesenteric, sacral, and cervical lymph nodes.164,165,166 Ltβr –/– mice have a phenotype similar to that of Ltα-/- mice.167 LTβR is also recognized by LTβ-related ligand, LIGHT, which also binds to the herpes virus entry mediator. LIGHT-LTβR signaling does not appear to play an essential role during lymphoid organogenesis, as no significant defect is observed in Light-/- mice168; however, mice doubly deficient in LIGHT and LTβ have fewer mesenteric lymph nodes than mice deficient in LTβ alone, indicating a cooperative effect of the two LTβR ligands. Treatment of pregnant females with an inhibitory soluble protein of LTβR (LTβR and human IgG Fc fusion protein, LTβR-Ig) inhibits most lymph nodes in the developing embryos, depending on the time of administration. Mesenteric lymph nodes are not inhibited by this treatment. These studies indicate that individual lymph nodes differ in the nature and time of cytokine signaling during development.169 Several additional cytokine and chemokinereceptor pairs are crucial for lymphoid organogenesis. Mice deficient in IL-7 or IL-7R,170,171 or TRANCE or TRANCER, also called RANKL and RANK,172,173 exhibit defects in lymph node development. CXCR5- or CXCL13-deficient mice174,




Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
