Fig. 1.1
Histopathological features of pilocytic astrocytoma. (a) Field of tumor cells demonstrating increased cellularity, mild nuclear atypia, and lack of mitoses. (b) Tumor edge with gliotic border (left of image) and neovascularization. (c) Biphasic pattern of compact, fibrillated astrocytes and loosely textured microcysts with a focus of endothelial proliferation. (d) Squash preparations demonstrating thin glial processes (“pili”) extending from bipolar tumor cells
Other grade I astrocytoma, glioma, and glioneuronal histologies that are seen in pediatric patients include subependymal giant cell astrocytoma, ganglioglioma, dysembryoplastic infantile ganglioglioma, and dysembryoplastic neuroepithelial tumor (Sievert and Fisher 2009).
Grade II astrocytomas are distinct from pilocytic tumors because of their location, degree of infiltration, and presence of genetic aberrations (Kleihues et al. 1993; Louis et al. 2007). Grossly, grade II astrocytomas are ill-defined lesions that tend to enlarge and distort involved structures. Destruction of brain tissue, however, is more characteristic of higher-grade tumors. Microscopic examination of resected grade II tumor specimens invariably shows diffuse infiltration of the surrounding gray and white matter. Low-power microscopy may show a subtle increase in overall cellularity and disruption of the orderly pattern of glial cells along myelinated fibers. Higher-power examination reveals neoplastic astrocytes with indistinct cytoplasmic features. The diagnosis is often based on the appearance of the nuclei, which are characteristically elongated. Nuclear atypia is minimal in low-grade astrocytomas and mitotic activity is infrequent.
1.2.2.2 Other Low-Grade Subtypes
Low-grade astrocytomas can be further subdivided on the basis of their microscopic appearance. The prognostic value of these subgroups is not entirely clear. Fibrillary astrocytoma is the most common grade II astrocytoma subtype and demonstrates a uniform, compact arrangement of fibrillary astrocytes with varying degrees of cellular atypia on a background of loosely structured tumor matrix (Steinbok and Mutat 1999). Gemistocytic astrocytomas are composed of neoplastic astrocytes with abundant eosinophilic, glial fibrillary acidic protein (GFAP)-positive cytoplasm with nuclei displaced to the periphery (Kaye and Walker 2000). The WHO classification identifies the gemistocytic subtype as low-grade astrocytoma, as long as cellularity and nuclear atypia remain mild (Louis et al. 2007). The pleomorphic xanthoastrocytoma (PXA), is a rare, GFAP-positive, astrocytic tumor typically occurring in the cerebral hemispheres of children and young adults (Kepes et al. 1973).
Histologically, PXA is characterized by large, neoplastic astrocytes with substantial nuclear pleomorphism and very atypical nuclei. The borders are often infiltrative, and tumor cells may display clustering in an epithelioid fashion (Lindboe et al. 1992; Powell et al. 1996). Desmoplastic infantile astrocytoma (DIA) is a rare tumor occurring in infants 18 months or younger. These tumors are usually large, cystic, supratentorial in location, and have a dural attachment. Histologically, they are loose to dense collagenous stroma with wavy fascicles of spindle cells (Taratuto et al. 1984). The rarest subtype is the protoplasmic astrocytoma, which has prominent microcysts, mucoid degeneration, and a paucity of GFAP positivity (Kaye and Walker 2000). Some consider this a histological pattern of fibrillary astrocytoma, rather than a true variant. Diffuse cerebellar astrocytomas resemble low-grade astrocytomas of the cerebral hemispheres with poorly circumscribed borders and invasion of the surrounding parenchyma. These tumors generally occur in older children, and young adults can undergo malignant transformation (Burger et al. 2000). Regardless of subtype, all low-grade astrocytomas have low cellularity, limited nuclear atypia, and rare mitotic activity. Low-grade astrocytomas with single mitotic figures have prognoses similar to other low-grade tumors (Giannini et al. 1999). A single mitotic figure suggests that the presence of isolated mitoses may not be sufficient to transform an otherwise low-grade astrocytoma to a higher-grade lesion.
1.2.2.3 Biology
Astrocytoma cytogenetic abnormalities occur less frequently and with different patterns in children than in adults (Cheng et al. 1999). In adult low-grade astrocytomas, mutations in the p53 tumor suppressor gene are common and may herald an early event in malignant progression (Watanabe et al. 1998; Kosel et al. 2001). In contrast, p53 mutations are not frequently found in the pediatric population (Litofsky et al. 1994; Felix et al. 1995; Ishii et al. 1998). The majority of pediatric pilocytic astrocytomas demonstrate normal cytogenetic findings (Griffin et al. 1988; Karnes et al. 1992; Bigner et al. 1997). In a recent study of 58 pediatric patients, 70 % of grade I astrocytomas had a normal cytogenetic profile (Roberts et al. 2001). In another study of 109 pediatric brain tumors, which included 33 low-grade astrocytomas, low-grade astrocytomas mostly showed changes in chromosome copy number (Neumann et al. 1993). Reported cytogenetic abnormalities include gains on chromosomes 1, 7, and 8 and losses of 17p and 17q (White et al. 1995; Wernicke et al. 1997; Zattara-Cannoni et al. 1998).
High-density single-nucleotide polymorphism-based genotyping and comparative genome hybridization (CGH) have revealed duplication or gain in chromosomes 5 and 7, with particular amplification of 7q34 in PA (Pfister et al. 2008; Sievert et al. 2008). Using CGH, BRAF was duplicated in 28 of 53 JPAs. In vitro inhibition of BRAF signaling, directly by lentivirus-mediated transduction of BRAF-specific shRNAs or indirectly by pharmacological inhibition of MEK1/2, the immediate downstream target of BRAF, caused G2/M cell-cycle arrest in astrocytic cell lines (Pfister et al. 2008). The amplification of 7q34 represents a duplication of the BRAF gene and fusion with the KIAA1549 gene. This BRAF–KIAA1549 fusion results in constitutively activated BRAF signaling, with subsequent downstream effects on cell proliferation and survival via MEK and ERK. The BRAF–KIAA1549 fusion transcript is detected in the majority of cerebellar pilocytic astrocytomas and less frequently in pilocytic astrocytoma in other locations as well as other low-grade glioma variants. Alternative Ras/Map kinase activating genetic changes have also been described in both pilocytic astrocytoma and other pediatric low-grade glioma histologies. The most common of these is the BRAF V600E mutation, described in 10 % of pediatric gliomas, as well as less commonly observed alternate fusion genes involving RAF (Chen and Guttman 2014; Gajjar et al. 2015). Thus, aberrant activation of the mitogen-activated protein kinase (MAPK) pathway, due to gene duplication or activating mutation of BRAF, is a common event in the tumorigenesis of pediatric low-grade astrocytomas and provides an opportunity for biologically targeted therapies with BRAF and/or MEK inhibitors.
Constitutive activation of the mTOR pathway is observed in pediatric low-grade glioma, through different mechanisms, in patients who develop either spontaneous or NF1-deficient PA (Dasgupta et al. 2005; Sharma et al. 2005). In tumors with Ras pathway-activating genetic lesions, mTOR, a downstream effector of the Ras pathway, is likely activated by upstream Ras activation (Chen and Guttman 2014). In patients with tuberous sclerosis-associated SEGA, mTOR is shown to be constitutively activated and responsive to treatment with mTOR inhibitors in the clinical setting (Ouyang et al. 2014; Franz et al. 2006, 2014). The identification of these markers may not only direct us to novel molecular targets for drug therapy, but may also allow rapid pathologic characterization and classification of these tumor types.
1.2.3 Clinical Features
Symptoms and signs caused by low-grade gliomas depend on the anatomic location, biological nature of the tumor, and age of the patient. These signs and symptoms may be nonspecific, such as those associated with increased intracranial pressure (ICP), or focal, related to tumor location. Nonspecific symptoms include headache, nausea, and vomiting, subtle developmental delay, and behavioral changes. Some of the behavioral changes associated with slow-growing tumors in children include alterations in personality, irritability, altered psychomotor function, apathy, and declining school performance. It is not uncommon for symptoms to have been present for months or years prior to diagnosis. In infants with open cranial sutures, a tumor may reach a massive size with a gradual increase in head circumference without signs of increased ICP or any other symptoms. Focal symptoms depend upon the location of the tumor and may include hemiparesis, monoparesis, hemisensory loss, dysphasia, aphasia, and impairment of recent memory. Tumors involving the optic pathways can present with quadrantanopia, homonymous hemianopsia, or, in cases with bilateral occipital lobe involvement, cortical blindness. Hemorrhage rarely occurs in low-grade tumors, although one report noted the presence of hemorrhage in 8 % of patients with pilocytic astrocytoma (White et al. 2008).
Epilepsy is a major presenting feature of pediatric patients with brain tumors, and seizures occur in more than 50 % of children with hemispheric tumors (Keles and Berger 2000). The majority of patients with tumor-associated epilepsy harbor slow-growing, indolent neoplasms such as low-grade gliomas. Other relatively slow-growing tumors, for example, astrocytomas, gangliogliomas, and oligodendrogliomas, may also present with a history of generalized seizures. Rapidly growing lesions are more likely to produce complex partial motor or sensory seizures, although generalized tonic-clonic seizures are also common.
1.2.4 Diagnostic Imaging
Magnetic resonance imaging (MRI) and computed tomography (CT) are essential tools in the diagnosis and treatment of brain tumors. Although CT is more commonly available, MRI provides higher sensitivity in differentiating tumor tissue from normal brain, allowing more detailed anatomic characterization of the lesion, and should be obtained in all children with a diagnosis of a brain tumor. A complete series should include the following sequences: T1-weighted axial and coronal (both before and after gadolinium), T2-weighted axial and coronal, and fluid-attenuated inversion recovery (FLAIR). In addition, sagittal plane sequences are helpful in defining anatomy of suprasellar and midline tumors. Other sequences such as fat suppression and MR angiography may also be required in specific situations. Newer techniques, such as magnetic resonance spectroscopy (MRS), functional MRI, and perfusion measurements, offer the potential of obtaining biochemical and functional information noninvasively (see Chap. 13). It is possible that in the future a pathologic diagnosis may be reached with substantial confidence without the need for open biopsy.
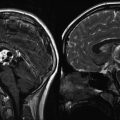
Although low-grade gliomas may produce considerable mass effect upon surrounding structures, neurologic deficits may be minimal. With the exception of pilocytic astrocytoma, low-grade astrocytomas are usually nonenhancing, iso- or hypodense masses on CT scan. Calcification may be detected in 15–20 % of cases, and mild to moderate inhomogeneous contrast enhancement can be seen in up to 40 % of all cases (Lote et al. 1998; Bauman et al. 1999; Roberts et al. 2000; Scott et al. 2002). Some tumors, characteristically PAs, may have cystic changes. On MRI, T1-weighted images show an iso- to hypointense nonenhancing mass that is hyperintense on T2-weighted images. Non-PA low-grade astrocytomas have minimal to no contrast enhancement following gadolinium administration (Fig. 1.2b, d). For this reason, the tumor boundary is difficult to determine with any T1-weighted sequence. FLAIR sequence is very sensitive for defining the extent of tumor infiltration (Fig. 1.2a, c).
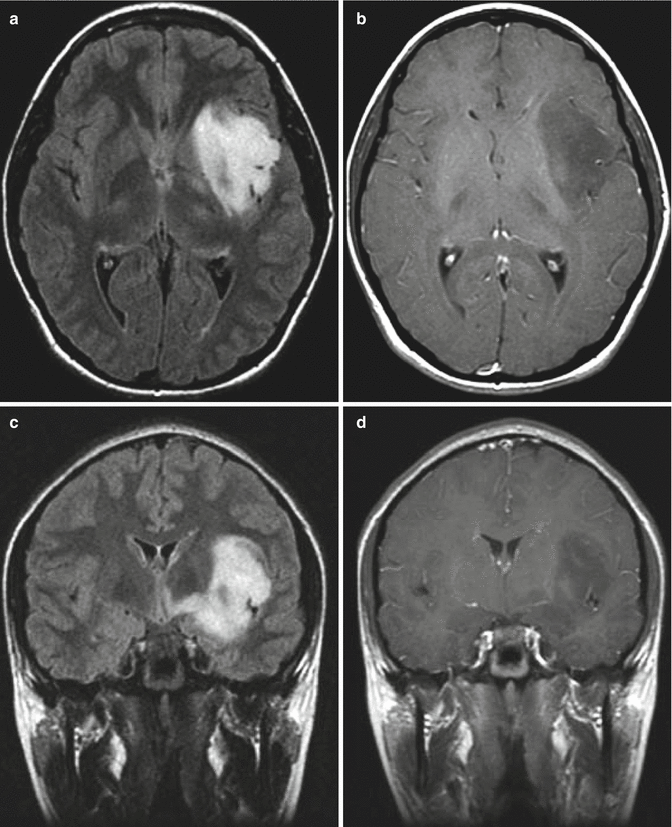
Fig. 1.2
MR images from a teenage girl with a low-grade astrocytoma of the insula who presented with a single seizure. Her neurologic exam was normal. (a, c) Axial and coronal FLAIR images showing the extent of involvement. Note the tumor infiltration medially under the lentiform nucleus toward the hypothalamus. (b, d) Corresponding T1-weighted post-gadolinium images showing no appreciable enhancement
Because many low-grade gliomas have a risk of progression or relapse after initial therapy, surveillance MR imaging over time is recommended typically at an interval of 3–6 months, depending on the degree of clinical concern for risk of relapse. In general, for grade II astrocytoma, the two most important features are an increase in the volume of T2-weighted FLAIR signal abnormality and/or new enhancement on post-gadolinium T1-weighted images. These features are also observed in patients who have received radiation treatment, and differentiating tumor recurrence from radiation necrosis continues to present a challenge. Additional information may be obtained from MR spectroscopy and positron emission tomography (PET) scans, but at times, the only method to confirm tumor recurrence is to obtain a surgical biopsy.
1.2.5 Treatment
1.2.5.1 Surgical Indications
A surgical procedure is usually the initial step in the management of low-grade gliomas. The primary objective is to obtain tissue for pathologic diagnosis. A relative exception would be for tumors in locations not amenable to surgery, such as optic pathway/chiasmatic gliomas, although a stereotactic biopsy can safely obtain tissue for histopathologic analysis. The secondary objective is to perform as extensive a resection as possible with acceptable neurologic outcome for the patient. The two variables that must be considered are the extent and timing of resection. Extent of resection is the most important prognostic factor for 5-year overall and progression-free survival (PFS). Patients who have partial resections or residual disease often recur or experience tumor progression (Shaw and Wisoff 2003; Kim et al. 2014). The feasibility of an open surgical approach depends upon several factors. The most important is the exact location of the tumor. Deep lesions within the basal ganglia, thalamus, motor cortex, or brainstem are usually not amenable to open surgical resection, while tumors in other locations can be accessed through various standard approaches. Other factors that modify the decision to attempt surgical resection are the patient’s clinical condition, age, associated hydrocephalus, and the surgeon’s assessment of risk of neurologic sequelae.
Timing of resection is a controversial topic, and few conclusive studies have been published to date. There are reports questioning the value of immediate treatment when an imaging study suggests a low-grade glioma, as no definitive evidence exists which demonstrates improvement in long-term survival following early intervention (Cairncross and Laperriere 1989; Recht et al. 1992).
In addition to reducing tumor burden and providing tissue diagnosis, resection permits management of increased ICP, prevention of irreversible neurologic deficits, decompression of adjacent brain structures, and control of seizures (Berger et al. 1991, 1993; Haglund et al. 1992; Keles and Berger 2000). For patients with discrete JPAs (WHO grade I), gross total resection (GTR), when possible, is curative. Contemporary neurosurgical methods, including ultrasonography, functional mapping, frameless navigational resection devices, and intraoperative imaging techniques enable more extensive resections with less morbidity.
1.2.5.2 Chemotherapy
Although indolent and slow growing, overall 5-year survival rates for patients with diencephalic and hemispheric tumors who have received radiation therapy vary, ranging from 40 % to 70 %. Additionally, the morbidity associated with radiation treatment can be substantial, prompting numerous investigators to explore chemotherapy as an alternative adjuvant treatment to control tumor progression. Chemotherapy effectively provides disease control in many optic pathway tumors (see below) and may improve prognosis for vision maintenance. Studies of early combination chemotherapy regimens with vincristine and actinomycin D, used in children less than 6 years of age, reported 62 % PFS without further therapy; those who did progress did so at a median of 3 years from the start of therapy. The median IQ in this group was 103 (Packer et al. 1988). It is important to recognize that prolonged periods of stable tumor size and clinical symptoms are considered a treatment “response” by many investigators. Alternative combination chemotherapy regimens have also resulted in tumor response in pilot studies. Other drug combinations that have been reported include lomustine and vincristine; 6-thioguanine, procarbazine, lomustine, and vincristine (TPCV); and combinations using cisplatin (Edwards et al. 1980; Gajjar et al. 1993). The combination regimen of carboplatin and vincristine (CV) has been associated with objective response rates (stable disease as well as tumor shrinkage) in the range of 60–70 % (Packer et al. 1997). The combination of TPCV has also been associated with a substantial response rate in a small cohort of patients (Prados et al. 1997).
A large-scale, randomized, phase III, multi-institutional clinical trial conducted by the Children’s Oncology Group (COG) examined the relative effectiveness of CV versus TPCV. Four hundred and one children less than 10 years old were enrolled in COG A9952. Of these 401 eligible children, 137 were randomized to receive CV, 137 were randomized to receive TPCV, and 127 patients with NF1 and radiographically verified progressive optic pathway glioma were nonrandomly assigned to the CV arm because of the heightened leukemogenic potential of TPCV in this patient population. Tumor response rates, defined as a decrease in both enhancement and T2 signal on MRI at the end of protocol therapy, were 57 % for CV, non-NF1; 61 % for CV, NF1; and 58 % for TPCV. The 5-year overall survival rates in CV-treated, non-NF1 versus NF1 patients were 86 % and 98 %, respectively. Similarly, 5-year event-free survival (EFS) was improved in NF1 versus non-NF1 patients (69 % vs. 42 %, respectively) and no difference in EFS was found when comparing CV versus TPCV. The median time to progression for CV versus TPCV was 3.2 versus 4.9 years (Ater et al.2012). A regimen of single-agent vinblastine demonstrated a 3- and 5-year EFS and OS of 43.2 % and 93.2 % (Bouffet et al. 2012). A phase 2 study of bevacizumab and irinotecan in patients with low-grade glioma demonstrated a 2-year PFS of 47.8 % (Gururangan et al. 2014). These findings demonstrate that both therapies can be used successfully to treat low-grade glioma with good overall EFS, thus allowing a delay in radiotherapy.
Although chemotherapy is documented to be active in low-grade glioma, conventional regimens are toxic and provide only transient tumor control. Investigators are exploring the role of mono- and combinatorial therapy to extend treatment response. The HIT-LGG 96 study examined the role of second-line chemotherapy in patients who had disease progression in the chemotherapy arm (94 patients). Of those 94 patients, 27 went on to receive a second round of chemotherapy consisting of vincristine/carboplatin and/or cyclophosphamide regimen, vinblastine alone, temozolomide alone, or other regimen. The median age in this group was 11.8 months. Best achievable response was tumor reduction in 8 patients and stable disease in 13 patients. Thirteen patients recurred 15.7 months after starting second-line chemotherapy. The overall 3-year PFS in the second chemotherapy group was 34 % (Kordes et al. 2008).
A phase II study assessed the efficacy of temozolomide in children with progressive optic pathway glioma and pilocytic astrocytoma. Thirty patients were treated with oral temozolomide for 5 days every 4 weeks. The 2-year PFS and overall survival rates were 49 % and 96 %, respectively, with manageable toxicity (Gururangan et al. 2007). These findings illustrate the potential to further delay radiotherapy in this pediatric population by using chemotherapy.
1.2.5.3 Radiation Therapy
As discussed above, low-grade astrocytoma may be curable with GTR. For those patients with unresectable or incompletely resected disease, the use of radiation therapy is controversial. There is some evidence to suggest that while radiation therapy may prolong PFS, it has little impact on overall survival (Pollack et al. 1995). Its use is largely limited to patients with progressive or recurrent disease or in the setting of a highly symptomatic patient who requires tumor stabilization to avert the progression of symptoms. A large-scale multi-institutional trial, SIOP-LGG 2004, sought to address the role of observation, adjuvant chemotherapy, and radiotherapy in order to assess their optimal therapeutic effect and toxicity on pediatric low-grade glioma after total or subtotal surgical resection. A total of 1,031 patients were enrolled and were nonrandomly assigned to one of three arms in an age-dependent manner. Six hundred sixty-eight patients were assigned to observation only, 216 to vincristine with carboplatin chemotherapy, and 147 to radiation/brachytherapy. Ten-year OS and PFS were 94 % and 47 %; three quarters of the chemotherapy-treated patients remain unirradiated with 9.3 years of median follow-up (Gnekow et al. 2012). In an 89 patient cohort of pediatric patients treated with conformal radiation for low-grade glioma at St. Jude’s Children’s Research Hospital, PFS and OS at 10 years were 75.3 % and 95.9 % (Merchant et al. 2009a, b). Eight-year PFS and OS in a cohort of LGG patients treated with intensity-modulated radiation therapy were 78.2 % and 93.7 %, with failures largely occurring in the tumor bed (Paulino et al. 2013). For the most part, these studies demonstrate the efficacy of radiation therapy in the treatment of pediatric low-grade gliomas. However, due to concerns about radiation-related side effects, an effort is generally made to delay or forgo radiation in young children.
Because of neurocognitive toxicity associated with radiotherapy, minimizing the dose and radiation fields using stereotactic radiosurgery or proton therapy may provide an effective alternative to standard conformal radiotherapy (Hadjipanayis et al. 2003; Marcus et al. 2005). One prospective trial using stereotactic radiosurgery demonstrated effective control of small, pediatric LGGs that had progressed either after surgery or chemotherapy. The 8-year PFS and overall survival rates using stereotactic radiosurgery in these patients were 65 % and 82 %, respectively (Marcus et al. 2005). Clinical outcomes using proton therapy in 32 pediatric patients treated for primary low-grade gliomas were comparable to standard radiotherapy. Neurocognitive exams posttreatment appeared stable, with minimal negative changes in working memory and processing speed, except in a subgroup of patients <7 years, who experienced significant declines in full scale IQ, as well as in patients who had significant dose to the left temporal lobe/hippocampus (Greenberger et al. 2014). Proton therapy appears to be safe and equally effective as IMRT or conformal therapy. Alternatively, the use of microsurgery combined with interstitial radiosurgical I-125 seed implantation (IRS) has demonstrated promising results. Nineteen children with low-grade glioma received IRS and/or microsurgery to the tumor site. With a median follow-up of 26 months, 5 tumors had a complete response, 11 tumors had reduction in size, two children developed radionecrosis requiring resection, and one child had progression and died (Peraud et al. 2008). In a cohort of pediatric patients treated with stereotactic brachytherapy, 10-year PFS and OS were 82 % and 93 %, respectively, again similar to other radiation strategies (Ruge et al. 2011). While this therapy appears feasible, long-term neurocognitive toxicity needs to be assessed.
1.2.5.4 Targeted Molecular Therapy
Overall prognosis and clinical outcome for patients with glioma are associated with tumor grade. Genes associated with glial cell grade and tumorigenesis continue to be identified. Understanding the pattern of genes activated in glioma will likely provide insight into the natural history and potential clinical course of these tumors and whether they will respond to standard chemotherapeutic regimens or novel molecular targeted therapies. For this reason, the PI3K/Akt/mTOR pathway has been studied in great detail as it plays a large role in the tumorigenesis of many cancers including glial tumors (Sabatini 2006; Guertin and Sabatini 2007).
Two complexes of mTOR exist: mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). The tumor suppressor genes TSC1/hamartin and TSC2/tuberin are important for the regulation of mTOR activity. Germline mutations of TSC lead to tuberous sclerosis and predisposition to a variety of benign tumors including hamartomas and lymphangioleiomyomas. Many upstream growth factor receptors and PI3K signal through the downstream mediator, mTOR. These observations make mTOR an attractive target for therapeutic intervention (Houghton and Huang 2004).
Further characterization of mTOR’s signaling pathway may lead to better application of mTOR inhibitor therapy. Franz et al. used rapamycin, an mTOR inhibitor, to treat 5 TSC patients who had either subependymal giant cell astrocytoma (n = 4) or pilocytic astrocytoma (n = 1). In all five cases, tumor regression was observed, and in one case, tumor necrosis occurred (Franz et al. 2006). As reviewed in Sect. 1.2.1.1.2, follow-up studies demonstrated very high response rates of TS-associated SEGA to the mTOR inhibitor everolimus. Based on these observations, as well as the role of mTOR signaling in sporadic and NF1-associated PA as reviewed in Sect. 1.2.2.3, inhibition of mTOR signaling is emerging as a provocative target for treatment of LGGs. In a cohort of 19 recurrent LGG patients treated with a combination of the EGFR inhibitor erlotinib and rapamycin, 1 patient had a partial response to treatment, and 6 patients had stabilization of disease for 12 months or greater (Yalon et al. 2013). A phase 2 study of treatment with everolimus alone in a cohort of 23 patients with recurrent or progressive low-grade glioma observed that 4 subjects had a partial response and 13 subjects had prolonged stable disease (Keiran et al. 2014).
Further exploration of gene expression profiles of grade I and II gliomas have already led to the introduction of novel therapies for pediatric low-grade gliomas. As reviewed in Sect. 1.2.2.3, BRAF is strongly implicated in the molecular pathogenesis of pediatric low-grade astrocytoma, and open a new avenue for molecularly targeted agents. In these studies, aberrant MAPK signaling could be inhibited in low-grade astrocytoma cell lines when treated with an inhibitor of the MAPK signaling component MEK. Initial efforts to treat low-grade glioma with the BRAF inhibitor sorafenib were disappointing, with 82 % of patients demonstrating uncharacteristically rapid progressive disease on treatment (Karajannis et al. 2014). Sorafenib was demonstrated to be associated with paradoxical activation of ERK in the setting of a BRAF–KIAA1549 fusion; hence, the drug may have driven tumor progression in this subset of patients (Sievert et al. 2013). In contrast, pediatric low-grade gliomas with BRAF V600E mutations have a high response rate to BRAFV600E-specific inhibitors such as dabrafenib, with 8 out of 15 patients having an objective radiographic response (Kieran et al. 2015). A preliminary report of a phase 1 study of the MEK inhibitor selumetinib (AZD6244) in pediatric low-grade glioma patients was notable for sustained responses in 8 of 38 patients treated, suggesting that MEK inhibition may be a promising therapeutic strategy for these patients (Banerjee et al. 2014), although a larger phase II study is currently underway that will hope to identify by genotype the patients most likely to respond to MEK inhibition. In summary, targeted therapies directed at the Ras/Map kinase pathway have shown significant early promise to treat pediatric low-grade gliomas, but it is still too early to determine which specific inhibitors (BRAF vs MEK) should be used to treat tumors with which particular mutation (NF1, BRAF fusion, BRAF V600E , RAS).
1.2.6 Outcome
Age and histological type are significant prognostic predictors. Although patients appear to benefit from more extensive resections, this issue remains controversial. In a majority of patients with tumor-associated epilepsy, including those patients with malignant astrocytomas, the seizures are infrequent and easily controlled with a single antiepileptic drug. In this setting, removal of the tumor alone usually controls seizure activity without the need for additional anticonvulsants. Children with indolent tumors, however, may have seizure activity that is refractory to medical therapy. Optimal seizure control without postoperative anticonvulsants in this situation is achieved when perioperative electrocorticographic mapping of separate seizure foci accompanies tumor resection. When mapping is not utilized, and a radical tumor resection includes adjacent brain, the occurrence of seizures will be lessened, but most patients will have to remain on antiepileptic drugs (Berger et al. 1991).
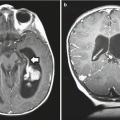
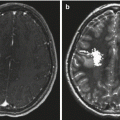
Dedifferentiation or malignant transformation is a well-described phenomenon in low-grade gliomas (Fig. 1.3). The incidence of recurrence as a higher histological grade ranges from 13 % to 86 % of tumors initially diagnosed as low grade (Keles et al. 2001). Similar to its broad range of incidence, the time to malignant differentiation is also variable, ranging from 28 to 60 months. However, factors resulting in change to a malignant phenotype remain unclear. In a recent study investigating the relationship between anaplastic transformation and patient’s age, a strong inverse relationship was found between age at initial diagnosis and time to progression to a higher-grade glioma (Shafqat et al. 1999).
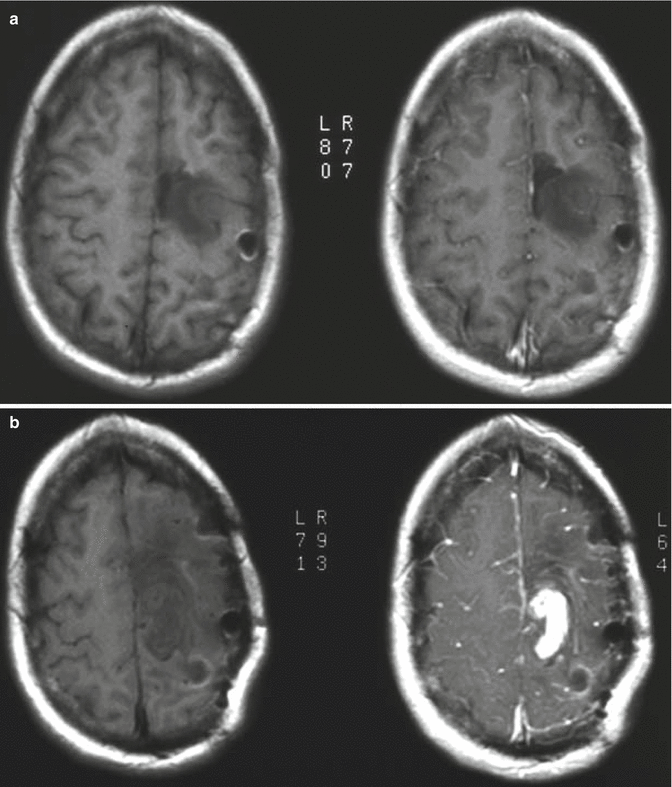
Fig. 1.3
Low-grade astrocytoma can recur at a higher grade. (a) Initial MRI demonstrates a nonenhancing mass in the left parietal lobe. Pathology was consistent with grade II astrocytoma. (b) Five years later, follow-up imaging demonstrates a new area of enhancement posterior to the original tumor. Pathology of the enhancing component was consistent with glioblastoma multiforme
In both low- and high-grade astrocytomas, the extent of surgical resection appears to correlate with outcome and quality of life (Pollack et al. 1995; Campbell and Pollack 1996; Keles et al. 2001; Wolff et al. 2002). Patients with GTRs live longer than those with partial resections, who in turn live longer than those who have biopsies only. A further consideration is that partial resection is often accompanied by significant postoperative edema surrounding residual tumor tissue, along with increased neurologic morbidity. However, the literature regarding the prognostic impact of surgery is controversial due to a lack of randomized studies addressing the issue. An additional complicating factor is the inconsistent and less subjective methodology used in determining the extent of resection. Historically, PFS at 3 years ranges from 61 % to 75 % for patients with low-grade gliomas (Packer et al. 1997; Gururangan et al. 2002). These patients have a 10-year survival rate of 70–90 %. More recently, however, data from the Surveillance, Epidemiology, and End Results (SEER) database reported on the long-term outcome of 4,040 children with low-grade glioma. Twenty-year overall survival was 87 %, and the 20-year cumulative incidence of death due to glioma was 12 %. Prognostic features included year of diagnosis, age at diagnosis, histology, WHO grade, primary site, radiation, and degree of initial resection in univariate analysis. In multivariate analysis, the greatest risk of death was associated with the use of radiation (Bandopadhayay et al. 2014).
1.3 Cerebellar Astrocytoma
Although astrocytomas as a group represent the most common tumor of the CNS in childhood, cerebellar astrocytomas comprise 10–20 % of all pediatric brain tumors (Lapras et al. 1986; Rutka et al. 1996; Smoots et al. 1998; Reddy and Timothy 2000) and 20–40 % of all posterior fossa tumors in children (Lapras et al. 1986; Rutka et al. 1996; Morreale et al. 1997; Steinbok and Mutat 1999; Reddy and Timothy 2000; Viano et al. 2001). Infratentorial tumors comprise approximately 50 % of all intracranial tumors in childhood and include medulloblastoma/PNET (20 % of the total), cerebellar astrocytomas (15 %), ependymoma (5 %), brainstem glioma (3 %), and other miscellaneous types (5 %) (Pollack 1999). Long-term survival after surgical resection is very high, but is dependent on histological type, extent of invasion, and completeness of tumor removal.
Recent laboratory investigations are attempting to define the molecular features of different grades of cerebellar astrocytomas. Clinical studies have focused on approaches to the treatment of residual/recurrent tumor, the role of adjuvant therapy, functional outcomes after treatment, and the management of complications, such as pseudomeningocele, cerebrospinal fluid (CSF) shunting, and cerebellar mutism.
1.3.1 Epidemiology
The incidence of cerebellar astrocytoma is difficult to determine accurately, but is estimated to be 0.2–0.33 cases per 100,000 children per year (Berger 1996; Gjerris et al. 1998; Rosenfeld 2000). The incidence peaks between ages 4 and 10 years, with a median age at diagnosis of 6 years (Steinbok and Mutat 1999). Twenty percent of these tumors occur in children less than 3 years of age (Rickert 1998). Gender does not play a role in disease predominance, prognosis, or survival (Rickert and Paulus 2001; Viano et al. 2001). International studies do not demonstrate a geographic or ethnic propensity for the occurrence of cerebellar astrocytomas, unlike craniopharyngiomas and germ-cell tumors (Gjerris et al. 1998; Rickert 1998; Rickert and Paulus 2001).
The term “cerebellar astrocytoma” has become synonymous with a benign tumor, although a small subset are high grade and malignant. The majority (80 %) of cerebellar astrocytomas in children are PAs (WHO grade I) and demonstrate a benign histology (Morreale et al. 1997). Fibrillary astrocytomas (WHO grade II) comprise 15 % of the total, while anaplastic astrocytomas (WHO grade III) and glioblastoma (GBM, WHO grade IV) each represent less than 5 % of the total (Steinbok and Mutat 1999). In patients who present with NF1, about 5 % will develop cerebellar JPAs (Li et al. 2001).
1.3.2 Pathology
1.3.2.1 Gross Appearance
Grossly, cerebellar astrocytomas can be cystic and solid or have mixed features. JPAs (WHO grade I) are typically cystic tumors containing yellow-brown fluid and neoplastic mural nodules. The cyst wall may contain either neoplastic cells or a pseudocapsule of glial tissue (Steinbok and Mutat 1999; Bonfield and Steinbok 2015). This classic appearance occurs in less than 50 % of cases. Diffuse subtypes are almost always solid tumors composed of circumscribed neoplastic cells without evidence of cysts. Very commonly, however, cerebellar astrocytomas demonstrate mixed appearance and consist of both cystic and solid portions of tumor. Cystic lesions tend to occur in the cerebellar hemispheres, while solid tumors often arise in the midline near the vermis and potentially extend to the brainstem (Abdollahzadeh et al. 1994).
1.3.3 Clinical Features
The mean age at diagnosis for cerebellar astrocytomas in children is 6.8 years, and the average duration of symptoms is 3–5 months (Steinbok and Mutat 1999; Reddy and Timothy 2000; Bonfield and Steinbok 2015). The slow-growing, indolent characteristics of these tumors allow functional compensation of adjacent brain tissue, and most cerebellar astrocytomas tend to be large at time of diagnosis. With greater availability of high-resolution neuroimaging, detection of these lesions is occurring earlier than in the past. Attempts to correlate age at diagnosis and prognosis have been inconclusive, and though patients diagnosed at younger ages tend to have better outcomes, more of these tumors tend to have a benign pathology (Morreale et al. 1997).
Initial signs and symptoms are usually mild and nonspecific and are caused by increased intracranial pressure. Headache is the most common presenting complaint (75–97 %) (Abdollahzadeh et al. 1994; Berger 1996; Steinbok and Mutat 1999; Viano et al. 2001; Bonfield and Steinbok 2015) and frequently occurs with recumbency. Decreased venous return and hypoventilation during sleep and recumbency exacerbate raised ICP (Steinbok and Mutat 1999). Headaches begin frontally and may migrate to the occiput. Constant occipital headache and neck pain with hyperextension are ominous signs of tonsillar herniation into the foramen magnum. Respiratory depression, preceded by cluster or ataxic breathing, may follow shortly (Rosenfeld 2000). Vomiting, found in 64–84 % of patients, is the second most frequent presenting symptom and is also caused by hydrocephalus and raised ICP (Steinbok and Mutat 1999; Viano et al. 2001). Papilledema occurs in 40–80 % of patients along with cerebellar dysfunction (Rashidi et al. 2003). In the absence of tumor infiltration of the area postrema, vomiting is usually not accompanied by nausea, unlike ependymomas and other lesions arising from the fourth ventricle itself.
Signs of cerebellar dysfunction include ataxia (88 %), gait disturbance (56 %), appendicular dysmetria (59 %), and wide-based gait (27 %) (Abdollahzadeh et al. 1994; Pencalet et al. 1999; Steinbok and Mutat 1999; Viano et al. 2001). Lesions of the cerebellar hemisphere produce ataxia and dysmetria in the ipsilateral limbs, while midline lesions produce truncal and gait ataxia (Berger 1996). Other clinical features include behavioral changes (32 %), neck pain (20 %), and papilledema (55–75 %) (Abdollahzadeh et al. 1994; Steinbok and Mutat 1999; Bonfield and Steinbok 2015). Some degree of hydrocephalus occurs in 92 % of cases, while seizures are extremely rare (2–5 %) (Abdollahzadeh et al. 1994). Cranial nerves and descending motor tracts are usually not affected, unless there is significant tumor extension, and involvement indicates probable brainstem infiltration. The only clinical feature related to poor prognosis is the presence of brainstem dysfunction (level of consciousness, motor-tract signs) regardless of histology (Sgouros et al. 1995).
1.3.4 Natural History
Cerebellar astrocytomas were once considered congenital posterior fossa brain tumors, requiring treatment only when symptomatic. Patients would typically report long-standing headaches and emesis, with occasional periods of relief. Patients with cerebellar symptoms often developed symptoms of syringomyelia, indicating unrelieved hydrocephalus. It was commonly believed that the cyst wall and cyst fluid were the cause of the patients’ symptoms. Thus, early treatment consisted of cyst-fluid decompression and cyst-wall removal. Symptoms were relieved temporarily, but patients often returned within months to years with cyst recurrence and sometimes tumors with malignant progression. Not until Cushing reported his surgical experience with 76 cerebellar astrocytomas in 1931 did it become clear that the true pathology lay in the mural nodule. If left untreated, patients would experience increasing bouts of cerebellar fits, become blind, and ultimately succumb to coma and death.
1.3.5 Diagnosis and Neuroimaging
1.3.5.1 Computed Tomography and Magnetic Resonance Imaging
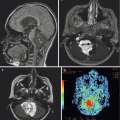
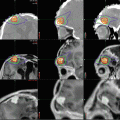
The classic radiographic appearance of a PA, observed in 30–60 % of cases, is a large cyst with a solid mural nodule (Fig. 1.4) localized to one of the cerebellar hemispheres (Steinbok et al. 1996; Reddy and Timothy 2000; Bonfield and Steinbok 2015). On CT, the cyst is hypodense to brain and hyperdense to CSF due to its high protein content, while on MRI, the cyst appears hypointense to brain on T1-weighted images and hyperintense on T2-weighted images. The mural nodule is hypo- to isodense to brain on CT and hyperintense to brain on T1-weighted images. The mural nodule enhances uniformly following contrast administration on both CT and MRI, while the cyst is not affected by contrast. The cyst wall, however, may demonstrate contrast enhancement if neoplastic cells are present (Fig. 1.4). In certain cases, the compressed glial reactive tissue surrounding a cyst may also show limited enhancement (Fig. 1.5). Other variations include multiple mural nodules; a single, large nodule filling in a portion of the cyst; and/or an irregular cyst contour.
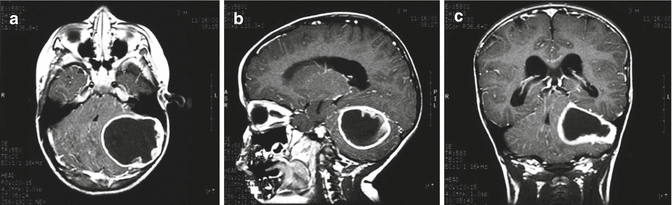
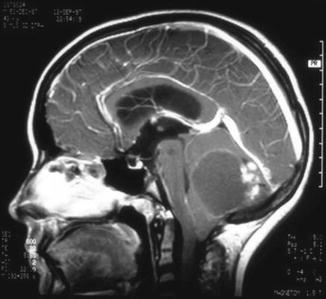
Fig. 1.4
Magnetic resonance (MR) images of a typical pilocytic cerebellar astrocytoma. (a) Axial, (b) sagittal, and (c) coronal T1-weighted MR images with gadolinium contrast demonstrating a cystic hemispheric lesion with mural nodule. In this case, the cyst wall enhances brightly following gadolinium and does represent tumor
Fig. 1.5
Sagittal magnetic resonance image of a cerebellar astrocytoma showing an irregular enhancing nodule located posterior to a large cyst. The cyst wall appears to enhance slightly, but this represents gliotic brain tissue
Cerebellar astrocytomas can also appear as solid lesions in 17–56 % of cases, with 90 % arising from or involving the vermis (Pencalet et al. 1999; Reddy and Timothy 2000). The CT shows a lesion hypo- to isodense to brain, and MRI demonstrates a solid mass hyperintense to brain. The solid tumor enhances uniformly following contrast administration in the majority of cases, but variations include regions of nonenhancement and small intratumoral cysts in up to 30 % of solid tumors (Campbell and Pollack 1996). Quite often, cerebellar astrocytomas will appear with both cystic and solid features and may have a rind-like enhancement pattern with varying degrees of cyst formation. Brainstem involvement is seen in 8–30 % (Steinbok and Mutat 1999; Reddy and Timothy 2000; Viano et al. 2001; Bonfield and Steinbok 2015) of cases, while the cerebellar peduncles are affected in 34 % (Hayostek et al. 1993; Pencalet et al. 1999). Calcifications are present in 10–17 % of tumors and hemorrhage in only 4.5 % (Berger 1996). Edema may be evident in some cases, but does not indicate malignancy or poor prognosis.
1.3.5.2 Magnetic Resonance Spectroscopy
Unfortunately, neither classic tumor appearance nor location on neuroimaging can confidently distinguish cerebellar astrocytoma from PNET or ependymoma. Biopsy with histological examination is necessary to establish a definitive diagnosis. Recently, MRS has been used to distinguish various pediatric cerebellar tumors based on differential levels of tumor metabolites and macromolecules. Pilocytic astrocytomas demonstrate increased choline:N-acetyl-aspartate (CHO:NAA) ratios and elevated lactate levels when compared to normal brain, similar to many other tumor types (Wang et al. 1995; Hwang et al. 1998; Warren et al. 2000). In one study, low-grade astrocytomas had higher NAA:CHO ratios than PNETs, but lower ratios than ependymomas, while creatine:CHO ratios were highest for ependymoma and lowest for PNET (Wang et al. 1995; Hwang et al. 1998). MRS may be useful to identify posterior fossa tumors in children after initial CT or MRI scanning. Other metabolites differentially detected in PNET and astrocytoma in vitro include glutamate, glycine, taurine, and myoinositol. One study examined subtotally resected low-grade astrocytomas and reported that higher normalized CHO levels significantly related to tumor progression 2 years following resection (Lazareff et al. 1998). Alternatively, high levels of lactate in pilocytic astrocytoma carry no indication of malignancy and may reflect aberrant glucose utilization in these tumors (Wang et al. 1995).
1.3.6 Treatment
1.3.6.1 Preoperative Management
Preoperative management depends on the clinical presentation of the patient. An asymptomatic, incidentally discovered lesion can be treated with an elective surgical intervention. More commonly, patients present with signs of increased ICP and cerebellar dysfunction and warrant urgent intervention. High-dose dexamethasone can relieve headache, nausea, and vomiting within 12–24 h and allow for several days of relief prior to a surgical procedure. An initial loading dose of 0.5–1.0 mg/kg given intravenously followed by a dose of 0.25–0.5 mg/kg/day divided every 6 h is the typical regimen (Rosenfeld 2000). In a patient who is stuporous and lethargic, with cardiorespiratory instability, relief of elevated ICP is of utmost importance and should be performed immediately. This is done by placing an external ventricular drain. In less urgent situations, an endoscopic third ventriculocisternostomy (ETV) can also be considered (Sainte-Rose et al. 2001). This procedure consists of placing a fenestration in the floor of the third ventricle to allow CSF to bypass an obstructive lesion in the posterior fossa. The ETV, although not always successful, can avoid permanent shunt placement. Currently, most surgeons will promptly proceed with tumor resection in the hope that relief of the obstructing mass will also treat associated hydrocephalus.
Ventriculoperitoneal (VP) shunting has been shown to improve survival after surgical resection of posterior fossa tumors. This procedure carries the risk of upward herniation and subdural hematoma from overshunting, while also rendering the patient shunt dependent for life with all of its associated complications. The risk of upward herniation is estimated at 3 % and presents with lethargy and obtundation around 12–24 h after shunt placement, with the potential for compression of the PCA at the tentorial hiatus, causing occipital lobe ischemia (Steinbok and Mutat 1999). Postoperative CSF diversion (with VP shunting) following complete tumor removal and unblockage of the aqueduct and fourth ventricle are required in 10–40 % of cases (Imielinski et al. 1998).
1.3.6.2 Surgical Treatment
GTR is the treatment goal and is achieved in 60–80 % of operative cases (Campbell and Pollack 1996; Gajjar et al. 1997). GTR is defined as the removal of all identifiable tumor tissue during surgery and is accomplished only when both the surgeon’s report and postoperative neuroimaging are concordant. An MRI with gadolinium enhancement is recommended within 24–48 h after resection. Postoperative changes, including swelling, edema, and gliosis appear by 3–5 days following surgery and may interfere with identification of residual tumor (Berger 1996). Residual tumor after GTR as noted by imaging is detected in 15 % of cases, while postoperative imaging fails to demonstrate known residual tumor as reported by the surgeon in 10 % of cases (Dirven et al. 1997). The clear presence of residual tumor is managed by reoperation to achieve complete resection.
Cystic tumors with a mural nodule may only require removal of the nodule to achieve complete resection, but removal of the cyst wall is dependent upon whether tumor is present. In some cases, contrast enhancement of the cyst wall on postcontrast MRI scans is clearly visualized (Fig. 1.4) and complete removal of all enhancing portions is considered essential to prevent recurrence. Nonenhancing areas do not require resection, and recent studies have shown that enhancement of the cyst wall does not always indicate tumor and may only represent vascularized reactive gliosis (Fig. 1.5) (Steinbok and Mutat 1999; Burger et al. 2000). There is also some evidence to suggest that patients who undergo complete cyst wall removal may have a poorer prognosis at 5 years than those with cyst walls left intact (Sgouros et al. 1995). Some support biopsy of the cyst wall during resection for frozen section; however, pathologic assessment is usually indeterminate and the sampling error is high, making biopsy of little value. Surgeons may choose conservative management of an enhancing cyst wall, especially if wall enhancement is thin (suggesting gliosis rather than tumor), biopsy samples do not demonstrate clear pathology, and gross appearance is benign (Steinbok and Mutat 1999).
Resection of cerebellar tumors can be associated with neurological deficits, although they are typically improved postoperatively (Steinbok et al. 2013). Subtotal resection (STR) is recommended when GTR would result in unacceptable morbidity and neurologic dysfunction, usually in the setting of brainstem invasion, involvement of the floor of the fourth ventricle, leptomeningeal spread, or metastasis. Involvement of the cerebellar peduncles was once thought to preclude GTR, but several authorities contend that GTR can be achieved in this circumstance (Berger 1996; Steinbok and Mutat 1999; Bonfield and Steinbok 2015), as postoperative deficits from resection involving the cerebellar peduncles tend to be transient. Management of incompletely resected tumors remains controversial and depends upon clinical circumstances.
1.3.6.3 Follow-Up Neuroimaging
Postoperative surveillance imaging in children with benign cerebellar astrocytomas depends on the extent of initial resection and the histology of tumor. While no standard schedule for surveillance imaging exists, large centers tend to obtain MRI scans at 3 and 6 months, then annually for 3–4 years. Routine imaging after confirmed GTR for a typical PA can be stopped 3–5 years following resection if there is no evidence of recurrence. However, due to the well-documented late recurrence behavior of a small percentage of benign cerebellar astrocytomas, sometimes decades after GTR, clinical changes should warrant reimaging. STR requires closer serial neuroimaging due to higher rates of tumor recurrence. Diffuse/fibrillary histology (grade II) is associated with STRs; however, GTRs of this histological subtype seem to demonstrate prognosis and recurrence rates rivaling those of juvenile pilocytic cerebellar tumors (grade I). Regardless of the extent of resection, most practitioners tend to follow grade II lesions more closely with serial exams and neuroimaging.
1.3.6.4 Management of Recurrence
Recurrence following GTR is rare and can occur after several years to decades from the initial operation. Reoperation with the goal of GTR is the recommended treatment for recurrence following STR, although this is usually not possible because the primary reason for incomplete resection is usually due to involvement of vital structures such as the brainstem (Akyol et al. 1992; Bonfield and Steinbok 2015). At reoperation, only 30 % of recurrences result in GTR, while 70 % continue to have residual tumor (Dirven et al. 1997). An interesting biologic feature of low-grade astrocytomas is the spontaneous regression or involution of residual tumors. For this reason, many authors advocate a period of observation for residual disease prior to reoperation. This approach is favored at our institution, particularly because a second procedure is associated with increased morbidity (Dirven et al. 1997).
Following STR, 30–40 % of patients have recurrence within 3 years (mean 54 months), while >60 % have recurrence by 5–6 years (Schneider et al. 1992). Tumors with diffuse/fibrillary histology are more prone to recurrence, but this association is not reported consistently in all series. Of all recurrent tumors, 65 % are pilocytic; 31 % are diffuse/fibrillary; 48 % are cystic; and 52 % are solid (Sgouros et al. 1995; Gjerris et al. 1998). Recurrences are found more often in the midline or vermis. Smoots et al., using multivariate analysis, noted that the only factor that predicted disease progression was the volume of residual disease (Smoots et al. 1998). This study also showed that only fibrillary histology, and not brainstem invasion or postoperative radiation therapy, significantly affects postoperative tumor volume. Unfortunately, the relationship between STR, brainstem invasion, residual tumor volume, and histology confound each other in almost all other series.
1.3.6.5 Adjuvant Therapy for Recurrence
Radiation therapy after resection plays an important role in the control of PNET and ependymoma, but its utility in cerebellar astrocytoma is incompletely understood. Postoperative radiation in subtotally resected tumors of any grade improves local control and recurrence rates, but survival rates seem to be unaffected (Garcia et al. 1990; Herfarth et al. 2001). One retrospective, nonrandomized study comparing patients with recurrence of grade I and II cerebellar astrocytoma found no significant difference in survival at both 5 and 9 years follow-up (Akyol et al. 1992). Radiation doses range from 30 to 54 Gy over 3–6 weeks, and some evidence suggests that doses greater than 53 Gy are necessary to see beneficial effects (Tamura et al. 1998; Herfarth et al. 2001). However, detrimental effects on the developing nervous system preclude its use in patients less than 3 years of age, and current trends favor delaying radiation therapy as long as possible to allow for maximal cognitive development prior to radiation therapy. The risks of radiation therapy include decreased cognitive function (Chadderton et al. 1995) and an increased risk of malignant transformation (Herfarth et al. 2001).
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


