Lipid, Apolipoprotein, and Lipoprotein Metabolism: Implications for the Diagnosis and Treatment of Dyslipidemia
Lipid, Apolipoprotein, and Lipoprotein Metabolism: Implications for the Diagnosis and Treatment of Dyslipidemia
Peter O. Kwiterovich Jr.
Human plasma lipoprotein metabolism is complex. An understanding of normal lipid, apolipoprotein, and lipoprotein metabolism is of paramount importance to provide a foundation from which one can appreciate the biochemical, genetic, and molecular basis of disorders of lipid and lipoprotein metabolism. Such understanding is also critical both for the proper selection of lipid-altering agents and for an interpretation of their efficacy.
Lipids are transported in plasma on lipoproteins, classified by their density and electrophoretic mobility into four major groups: chylomicrons, very-low-density (pre-β) lipoproteins (VLDL), low-density (β) lipoproteins (LDL), and high-density (α) lipoproteins (HDL) (Table 1.1). After electrophoresis, chylomicrons remain at the origin, and VLDL, LDL, and HDL migrate in the same positions as pre-β-, β-, and α-globulins, respectively. Chylomicrons and VLDL primarily transport triglycerides (TG) while LDL and HDL are cholesterol-rich lipoproteins.
Historically, the term “hyperlipidemia” was developed to refer to an elevation of plasma cholesterol or TG, or both. In 1967, Fredrickson et al. (1) translated hyperlipidemia into hyperlipoproteinemia by developing a lipoprotein classification system based on the electrophoretic migration of the four major lipoprotein classes. Plasma was applied to a paper strip, subjected to electrophoresis and the lipoproteins detected by a lipid stain. Chylomicrons remained at the origin, and were followed sequentially by VLDL, LDL, and then HDL. Hyperlipoproteinemia was due to elevations of chylomicrons alone (type I); LDL alone (type IIa); LDL plus VLDL (type IIb); VLDL plus VLDL remnants (type III); VLDL alone (type IV); and chylomicrons plus VLDL (type V) (see also Chapter 2). These lipoprotein patterns were really phenotypes reflecting different kinds of hyperlipoproteinemia, which could be primary or secondary to other causes such as hypothyroidism and renal disease. A given phenotype was not diagnostic for a genotype. The levels of the individual lipoproteins within a given phenotype were measured, using a combined heparin manganese precipitation/preparative ultracentrifugation method (see also Chapter 2). Lipoprotein patterns IIa, IIb, III, IV, and V were often associated with cardiovascular disease (CVD), while types I and V were commonly associated with pancreatitis. No lipoprotein pattern was described for HDL.
John Gofman is generally considered the father of the modernday lipoprotein field (2). Using analytical ultracentrifugation to characterize and quantify the major lipoproteins (and their subclasses) (see also Chapter 2), Gofman et al. (3) found in the prospective 10-year Livermore study in men that a low level of both major subclasses of HDL, that is, HDL2 and HDL3, significantly predicted an increased risk of CVD. Earlier, Barr et al. (4) had reported “low alpha lipoproteins” in patients with atherosclerosis, diabetes, or the nephrotic syndrome. Now, the importance of low HDL (also referred to as hypoalphalipoproteinemia [hypoalpha]) is well established as an independent predictor of CVD (5) (see also Chapter 9). In contrast, elevated levels of HDL (also referred to as hyperalphalipoproteinemia [hyperalpha]) predicted decreased CVD, and in some families longevity (6).
As well, it became appreciated that low levels of LDL (hypobetalipoproteinemia [hypobeta]) could occur, especially in families (7). In some kindreds, low LDL predicted decreased CVD and longevity (6). In a few others, LDL was undetectable, or very low, reflecting phenotypes of severe hypobeta that led to problems of intestinal fat absorption and neurologic problems (8) (see also Chapter 8).
Thus, the term hyperlipoproteinemia alone was insufficient to describe the multiple phenotypes of disorders of lipoprotein metabolism, and the phrase “dyslipoproteinemia” was born. Dyslipoproteinemia implies that the lipoproteins might be either elevated or low in their concentration and still be associated with disease. But dyslipoproteinemia further encompasses the clinical situation where the lipoproteins are normal, as judged by their cholesterol content, but there is still an underlying abnormality, such as an increased number of small dense LDL particles (hyperapobetalipoproteinemia [hyperapoB]) (see Chapter 8) or the presence of an elevated lipoprotein(a) or Lp(a) (see Chapter 18).
In this chapter and in this book, we have taken the poetic license to use the word dyslipidemia, even when it is understood that dyslipoproteinemia is technically more correct. Dyslipidemia is easier and faster to say. Just as “lipidologist” is easier than “lipoproteinologist.” Dyslipoproteinemia or dyslipidemia can result from the expression of a mutation in a single gene that plays a paramount role in lipoprotein metabolism. A myriad of defects have been described in the apolipoproteins, special amphipathic proteins that not only solubilize the nonpolar lipids in plasma but are critical for a number of other functions that I will review here. Defects can also occur in enzymes and carrier proteins that modify their normal effects on circulating lipoproteins. Molecular abnormalities also can be present in a number of receptors that facilitate the cellular uptake of intact lipoproteins or of a lipid that they may carry. Finally, there are defects in transporters in cell membranes that disrupt the normal uptake or egress of lipids to or from the cells.
From a practical standpoint, more often dyslipidemia reflects the influence of multiple genes. As well, environmental influences such as excessive dietary intake of fat and calories and limited physical activity, particularly when associated with overweight or obesity, can also contribute significantly to dyslipidemia. This chapter presents the theoretical basis for normal and abnormal lipoprotein metabolism and a discussion of the practical implications of such knowledge.
THEORETICAL CONSIDERATIONS
Lipoprotein Classification and Properties
Four Major Lipoprotein Classes
Plasma lipoproteins are spherical particles consisting of a core that contains nonpolar lipids, mostly TG and cholesteryl esters (CE), surrounded by a surface coat consisting of proteins (apolipoproteins), and more polar lipids, phospholipids (PL), and unesterified (free) cholesterol (FC). The hydrated density of the lipoproteins is related to their chemical composition and the relative content of lipid and apolipoprotein. Chylomicrons (< 0.95 g/mL) are 99% lipid, most of it being TG (Table 1.1). After plasma has stood overnight, these large particles (80 to 500 nm) will rise to the top, where they appear as a creamy layer. VLDL (0.95 to 1.006 g/mL) are about 90% lipid, the majority of it being TG, with lesser amounts of cholesterol. When present in plasma in increased amounts, VLDL can be large enough to impart a cloudy or turbid appearance to plasma. LDL (1.019 to 1.063 g/mL) are the major carriers of cholesterol in plasma, and about 50% of their weight is CE and FC. Even when LDL is markedly elevated by itself, the plasma remains clear. HDL (1.063 to 1.21 g/mL) comprise about equal amounts of apolipoprotein and lipid, principally PL and cholesterol.
TABLE 1.1 CLASSIFICATION AND PROPERTIES OF THE MAJOR HUMAN PLASMA LIPOPROTEINS
Transport intestinal triglycerides and cholesterol
Transport hepatic triglycerides and cholesterol
Provide cholesterol to cells
Reverse cholesterol transport
a Includes the mass of cholesteryl ester and esterified cholesterol.
Apolipoproteins
Apolipoproteins have amphipathic helices, allowing the nonpolar amino acid residues to interact with the lipid component of the lipoprotein, and the polar amino acids to interact with the aqueous environment of plasma. In addition to allowing lipids to be transported through plasma, apolipoproteins have other generic functions such as releasing lipoproteins from cells, facilitating the cellular uptake of intact lipoproteins, or selective uptake of their lipids, and regulating other aspects of lipoprotein metabolism such as intravascular remodeling and lipid transfer.
The major lipoproteins have a variety of apolipoproteins associated with them, each of which has one or more functions. The nomenclature for the apolipoproteins follows an alphabetical scheme (Table 1.2). Apolipoprotein A-I (apoA-I) constitutes about 70% of the apolipoproteins of HDL. Apolipoprotein B (apoB) is the major apolipoprotein of chylomicrons, VLDL, and LDL (Table 1.1), and of the lipoprotein particles that result from the hydrolysis of TG, namely, chylomicron remnants, VLDL remnants, and intermediate-density lipoproteins (IDL) (1.006 to 1.019 g/mL). ApoB is also a major component of Lp(a) that consists of one molecule of apoB joined through a disulfide bridge to apo(a), a glycoprotein homologous to plasminogen (see also Chapter 18). The full-length apoB polypeptide (MW 550,000), apoB-100, is made in the liver and is found in VLDL, VLDL remnants, IDL, LDL, and Lp(a). A truncated version of apoB, apoB-48, is made in the intestine and is found in chylomicrons and chylomicron remnants. ApoB-48 is the product of the same gene as apoB-100 but the MW of apoB-48 is 240,800 or 48% of that of apoB-100 (Table 1.2), due to a post-translational modification.
TABLE 1.2 CLASSIFICATION AND PROPERTIES OF HUMAN PLASMA APOLIPOPROTEINS
Apolipoprotein
Molecular weight
Chromosomal location
Function
ApoA-I
29,016
11q23
Cofactor LCAT; facilitates both the transfer of cell cholesterol by ABCA1 to nascent HDL and the delivery of CE and FC on HDL to liver through SR-BI
ApoA-II
17,414
1q21-23
Inhibits TG hydrolysis by HL and VLDL
ApoA-IV
44,465
11q23
Activates LCAT; promotes formation of chylomicrons
ApoA-V
39,000
11q23
Stimulates proteoglycan-bound LPL
ApoB-100
512,723
2p24-p23
Secretion of VLDL from liver; binding ligand to LDLR
ApoB-48
240,800
2p24-p23
Secretion of chylomicrons from intestine
ApoC-I
6,630
19q13.2
Inhibits apoE binding to LDLR; stimulates LCAT; inhibits CETP and SR-BI
ApoC-II
8,900
19q13.2
Cofactor LPL
ApoC-III
8,800
11q23
Noncompetitive inhibitor of LPL; inhibits binding of ApoE on TG-rich lipoproteins to LDLR
ApoD
19,000
3q26.2
Promotes reverse cholesterol transport
ApoE
34,145
19q13.2
Binding ligand for LRP on chylomicron remnants and for LDLR on VLDL and IDL
ApoH
54,000
17q23
Activates LPL
ApoL
42,000
22q13.1
Made only in the pancreas; affects TG and glucose metabolism but mechanism unknown
The apoB-containing lipoproteins (chylomicrons and VLDL and their remnants, IDL, LDL, and Lp(a)) are associated with increased risk from CVD. In contrast, the apoA-I-containing lipoproteins (HDL and their subclasses, HDL2 [1.063 to 1.120 g/mL]) and HDL3 [1.120 to 1.210 g/mL]) are generally associated with reduced risk of CVD.
Plasma lipoprotein classes are associated with a number of other apolipoproteins (Tables 1.1 and 1.2). The characteristics of these major apolipoproteins and some of their functions are summarized in Table 1.2. These functions will be exemplified when the pathways of lipoprotein metabolism will be discussed in the following text. The entire sequence of the gene for each apolipoprotein listed in Table 1.2 has been determined (http://www.ncbi.nlm.nih.gov/sites/entrez). A significant number of investigators have contributed to the exquisite knowledge of the apolipoproteins. Dr. Petar Alaupovic and colleagues also developed and proposed a classification system according to “lipoprotein families,” which were isolated from the major lipoproteins according to the presence or absence of certain apolipoproteins. Such differences had pathophysiologic consequences and provided further insight into the effect of apolipoproteins within each major lipoprotein class.
Lipoprotein Metabolism: Key Enzymes and Lipid Transport Proteins
There are four enzymes and two transfer proteins whose functions are critical to the regulation of lipoprotein metabolism (Table 1.3). Lipoprotein lipase (LPL), hepatic lipase (HL), and endothelial lipase (EL) are homologous lipolytic enzymes that hydrolyze TG into free fatty acids (FFA) and monoglycerol, and PL into lysophospholipids and FFA. They have different major substrates, namely LPL, chylomicrons, and large VLDL; HL, small VLDL, IDL, and large HDL2; and EL, large HDL2 (Figs. 1.1 and 1.2). The cofactor for LPL is apolipoprotein C-II (apoC-II). Lecithin cholesterol acyltransferase (LCAT) transfers a fatty acid from lecithin to FC in nascent HDL, forming CE and promotes the formation of a spherical HDL (Fig. 1.2). LCAT requires apoA-I as a cofactor. Phospholipid transfer protein (PLTP) transfers lipids from VLDL to HDL during lipolysis facilitating the formation of a larger HDL. Cholesteryl ester transfer protein (CETP) transfers TG from VLDL to LDL and HDL in exchange for CE. The TG on LDL and HDL can then be hydrolyzed by HL producing smaller, denser particles. The CE on LDL (and several other apoB-containing lipoproteins) are then removed by the LDL receptor (LDLR) in liver (Fig. 1.1).
TABLE 1.3 LIPOPROTEIN METABOLISM: KEY ENZYMES AND LIPID TRANSPORT PROTEINS
MW
Tissue of origin
Function
LPL
50,394
Heart, adipose, skeletal muscle
Hydrolyze TG and phospholipids of chylomicrons and large VLDL
HL
53,322
Liver (hepatocytes)
Hydrolyze TG and phopholipids of small VLDL, IDL and large HDL2
EL
55,000
Endothelial cells in liver, lung macrophages, testis, ovary
Catalyzes the hydrolysis of HDL phospholipids and facilitates HDL clearance
LCAT
47,090
Liver
Transfer of two acyl groups from lecithin to free cholesterol to form CE in HDL
CETP
74,000
Liver, adipose
Transfer TG from VLDL to LDL and HDL in exchange for CE
PLTP
81,000
Liver, lung, adipose
Transfer phospholipids and free cholesterol from VLDL to HDL during lipolysis promoting formation of large HDL
LPL, lipoprotein lipase; TG, triglycerides; VLDL, very low density lipoproteins; HL, hepatic lipase; IDL, intermediate density lipoproteins; EL, endothelial lipase; HDL, high density lipoproteins; LCAT, lecithin cholesterol acyltransferase; CETP, cholesteryl ester transfer protein; LDL, low density lipoproteins; PLTP, phospholipid transfer protein.
Lipoprotein Metabolism of ApoB-containing Lipoproteins: Major Receptors and Transporters
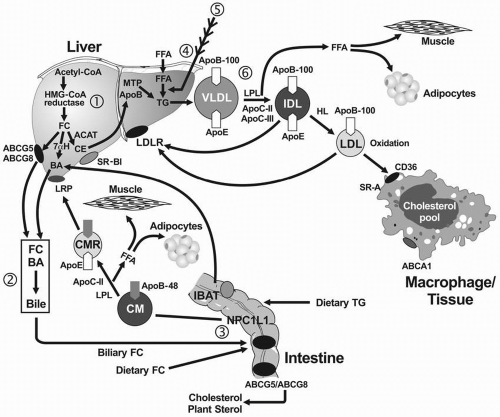
The transport of plasma lipids by apoB-containing lipoproteins may be conceptually divided into intestinal and hepatic systems (Fig. 1.1).
Intestinal Lipid Transport
Absorption of Lipids. Most dietary lipid is in the form of neutral fat or TG (75 to 150 g/day). The amount of cholesterol in the diet is usually about 300 mg/day, but varies from 100 to 600 mg/day. In addition to dietary cholesterol, about 1,100 mg of biliary cholesterol is secreted each day from the liver into the intestine (Fig. 1.1). In the small intestine, lipids are emulsified by bile salts and hydrolyzed by pancreatic lipases. The bile acids are then reabsorbed by the intestinal bile acid transporter (IBAT) in the ileum for return to the liver through the enterohepatic pathway (Fig. 1.1). TG is broken down into fatty acids and 2-monoglycerides; CE are hydrolyzed into FFA and unesterified FC. These components are then absorbed by the intestinal cells. The absorption of cholesterol occurs in the jejunum through the high-affinity uptake of dietary and biliary cholesterol by the Niemann-Pick C1 Like 1 (NPC1L1) protein (Fig. 1.1). Normally, about 50% of dietary and biliary cholesterol is absorbed daily, but the range of absorption is considerable (20% to 80%). Excessive sterol absorption is prevented by two ATP-binding cassette proteins, the ABCG5/ABCG8 transporters, which act together to pump excess cholesterol and plant sterols from the intestine back into the lumen for excretion into the stool (Fig. 1.1).
Formation, Secretion, and Metabolism of Chylomicrons. Inside the intestinal cells, monoglyceride is re-esterified to FFA forming TG and cholesterol is esterified by acyl cholesterol acyltransferase (ACAT). Both lipids are packaged into chylomicrons, along with apoB-48, apoA-I, and apoA-IV. Intestinal lipoprotein assembly is complex (9) and covered in more detail in Chapter 7. Chylomicrons are secreted into the thoracic duct from which they enter the peripheral circulation where they acquire apoC-II and apolipoprotein E (apoE) from HDL. Chylomicrons are too large to cross the endothelial barrier, and apoC-II, a cofactor for LPL, facilitates the hydrolysis of TG at the endothelial lining of blood vessels. The FFA that are released are taken up by muscle cells for energy utilization or by adipose cells for re-esterification into TG (Fig. 1.1). As a result, a chylomicron remnant is produced that is enriched in CE and apoE. This remnant is rapidly taken up by the liver by a process that involves an initial sequestration of remnant particles on hepatic cell surface proteoglycans, followed by receptor-mediated endocytosis of remnants through the interaction of apoE with the chylomicron remnant receptor (LRP) (also called the low density lipoprotein receptor-like protein) or with the LDLR on the surface of hepatic parenchymal cells (10) (Fig 1.1). The initial binding to proteoglycans may be facilitated by LPL, which possesses both lipid and heparinbinding domains (10).
The uptake of dietary and biliary cholesterol is part of a process that regulates the pool of hepatic cholesterol by downregulation of the LDLR and by inhibition of the rate-limiting enzyme of cholesterol biosynthesis, hydroxymethylglutaryl (HMG)-CoA reductase (see also the following text). Hepatic cholesterol can be secreted into bile unchanged via ABCG5/ABCG8, converted by 7α-hydroxylase into bile acids, or used for lipoprotein synthesis (Fig. 1.1).
Hepatic Lipid Transport
VLDL Synthesis. In the fasting state, most TG in plasma is carried by VLDL. The amount of TG, CE, and apoB-100 in liver is critical for VLDL synthesis. In the liver, FFA is normally activated (fatty acid CoA) and then oxidized or incorporated into TG or CE. When there is an increased flux of FFA to the liver, for example, from insulin-resistant adipocytes (see also Chapters 7, 8, and 10), the ability of the oxidative or storage pathways to metabolize fatty acid CoA is exceeded. Intermediates of fatty acid metabolism, such as diacylglyceride, accumulate and can both stimulate TG formation and activate different serine kinases that negatively regulate insulin action (11) (see also Chapter 10). This latter inhibitory effect on insulin action increases cholesterol synthesis and stimulates apoB secretion, since insulin normally decreases cholesterol synthesis and thereby inhibits apoB secretion.
ApoB-100 is constitutively made in the liver. The expression of the apoB gene is therefore not regulated up or down. The amount of apoB-100 in liver is regulated by proteolysis. For example, only some of the apoB-100 molecules that are made survive and become incorporated into VLDL; the remainders are degraded by proteolytic enzymes. As apoB-100 interacts with CE, it likely assumes a new conformation, leading to decreased degradation of apoB and thus to its increased production. TG is then incorporated into this complex through the action of microsomal transfer protein (MTP), producing VLDL. PLTP may also function in hepatocytes to add lipid to nascent apoB, thereby limiting apoB degradation and increasing VLDL production.
FIGURE 1.1 Pathways of exogenous (intestinal) and endogenous (hepatic) lipoprotein metabolism. CM transport dietary TG and cholesterol of dietary and hepatic origin. CE, FC, and TG are emulsified by BA, hydrolyzed by pancreatic lipases, absorbed and resynthesized, and packaged by MCT and apoB-48 into chylomicrons that are secreted. The TG in CM are hydrolyzed by LPL and apoC-II producing FFA, which are taken up by adipocytes or muscle. The resulting CMR is then removed by the LRP receptor, delivering CE and FC to liver. BA are reabsorbed through the IBAT and recycled to the liver. FC is synthesized in the liver through HMG-CoA reductase and can be excreted from the liver into bile by ABCG5/ABCG8, or converted to BA by 7αH, or esterified by ACAT into CE. CE interacts with apoB-100, reducing its proteolysis, and TG is added by MCT, producing VLDL, which contains one molecule of apoB-100 that is required for its secretion. The TG on VLDL are hydrolyzed by LPL and apoC-II, producing FFA and IDL. Some IDL is removed by the interaction of apoE with the hepatic LDLR, the rest is converted into LDL by HL. ApoC-III interferes with apoE-mediated IDL uptake. LDL is normally removed by the LDLR; excess LDL can be oxidized in the vascular wall and taken up by the scavenger receptors, CD36 and SR-A on macrophages, promoting CE storage. CM, chylomicrons; TG, triglycerides; CE, cholesteryl esters; FC, free cholesterol; BA, bile acids; MTP, microsomal triglyceride transport protein; LPL, lipoprotein lipase; FFA, free fatty acids; CMR, CM remnant; IBAT, intestinal bile acid transporter; HMG-CoA, hydroxymethylglutaryl-CoA; 7αH, 7α-hydroxylase; ACAT, acyl cholesterol acyltransferase; VLDL, very-low-density lipoprotein; IDL, intermediate-density lipoprotein; LRP, low density lipoprotein receptor-like protein; LDLR, low density lipoprotein receptor; LDL, low density lipoproteins; HL, hepatic lipase; CD36, macrophage scavenger receptor-B; SR-A, scavenger receptor A.
VLDL Secretion. ApoB-100 is necessary for the secretion of the TG-rich VLDL particles. Following the addition of CE and TG to apoB-100, and of other lipids and apolipoproteins (Table 1.1), such as PL, apoE, apolipoprotein C-I (apoC-I), apoC-II, and apoC-III, VLDL are secreted into plasma.
VLDL Catabolism. VLDL-TG are hydrolyzed by LPL and its cofactor apoC-II to produce VLDL remnants and then IDL, the smallest remnant (Fig. 1.1). Compared with VLDL, IDL are relatively enriched in CE and depleted in TG. Some IDL are taken up directly by the interaction of apoE with the LDLR on liver, while others are hydrolyzed by HL, to produce LDL, the final end product of VLDL catabolism (Fig. 1.1). TG can also be transferred from VLDL and IDL to HDL and LDL in exchange for CE by CETP (Fig. 1.2).
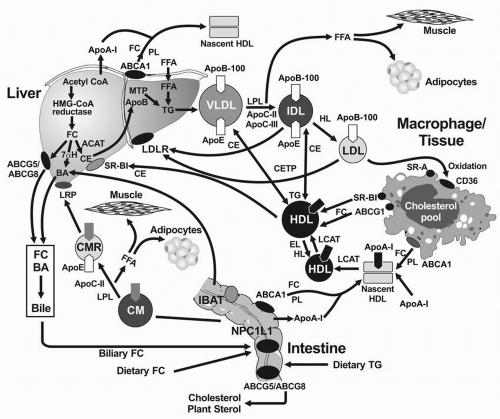
FIGURE 1.2 The reverse cholesterol transport pathway and its interaction with the exogenous (intestinal) and endogenous (hepatic) pathways. ApoA-I is synthesized and secreted by intestine and liver, after which it interacts with ABCA1, promoting the egress of FC and PL and the formation of the nascent cigar-shaped HDL particle. LCAT and apoA-I catalyze the formation of CE by adding an FFA from the PL to FC, producing a spherical HDL particle, which becomes larger through LCAT activity. Through the action of CETP, the large HDL exchanges some of its CE for TG from the apoB-containing lipoproteins, which are then removed by the LDLR. The CE on large HDL can also be delivered to the liver by specific uptake of the SR-BI receptor. Once inside the liver cholesterol must be excreted into bile directly through ABCG5/ ABCG8 or converted into BA by 7αH to complete the reverse cholesterol transport pathway. ApoA-I, apolipoprotein A-I; ABCA1, ATP binding casette transporter A1; FC, free cholesterol; PL, phospholipids; HDL, high-density lipoproteins; LCAT, lecithin cholesterol acyltransferase; CETP, cholesteryl ester transfer protein; CE, cholesteryl esters; FFA, free fatty acids; LDLR, LDL receptor; SR-B1, scavenger receptor class B1; ABCG5/ABCG8, ATP binding casette transporters G5 and G8; BA, bile acids; 7αH, 7α-hydroxylase.
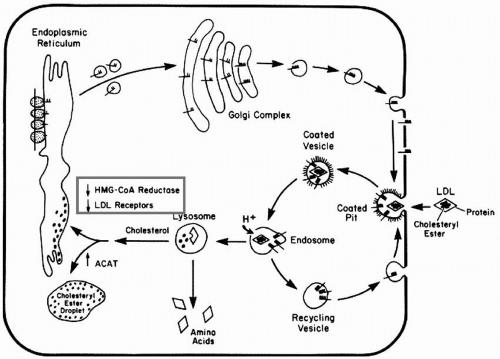
LDL Binding, Internalization, and Degradation. The LDLR pathway was elegantly elucidated by Brown and Goldstein (12) (Fig. 1.3) (see also Chapter 8). The LDLR is synthesized as a 120-kDa precursor. After glycosylation in the endoplasmic reticulum (ER) and Golgi, the mature 140-kDa LDLR reaches the cell surface where it is directed toward clathrin-coated pits (see Chapter 8) (Fig. 1.3). When the LDLR is in clathrin pits on the cell surface, its extracellular domain is extended, exposing the ligand-binding domain of the LDLR to LDL apoB-100. The receptor-ligand complex is internalized within coated vesicles by endocytosis and delivered to endosomes (Fig. 1.3). An adaptor protein called autosomal recessive hypercholesterolemia (ARH), after the disorder that permitted its discovery (see also the following text), couples the LDLR to the endocytic machinery, permitting LDLR internalization. In the acidic environment of the endosome, LDL is displaced from the LDLR, permitting release of LDL in the endosome and recycling of the LDLR to the cell surface. The released LDL is subsequently degraded in the lysosome. The apoB-100 moiety undergoes proteolysis, and CE are hydrolyzed into FC and FFA. The FC derived from LDL decreases hydroxymethylglutaryl-CoA (HMG-CoA) reductase and LDLR activity by inhibiting the sterol regulatory element binding protein (SREBP) pathway (see the following text).
In addition to the apoB-100 moiety on LDL, the LDLR also binds with high affinity to apoE on TG-rich lipoproteins, promoting their internalization and cellular metabolism. This process appears to occur independently of ARH (13).
Cholesterol regulates the proteolytic release of SREBP, a transcription factor, from the ER (12,14) (see also Fig. 8.3). This effect occurs through the SREBP cleavage activating protein (SCAP) that is both a sensor of sterols and an escort of SREBP (see also Chapter 8). For example, when hepatocytes are depleted of cholesterol, SCAP transports SREBP from the ER to the Golgi, where two proteases, Site-1 protease and Site-2 protease act in sequence to release the NH2 terminal of SREBP from the membrane (see also Fig. 8.3). The NH2 terminal of SREBP containing the bHLH-Zip domain of SREBP enters the nucleus and binds to a sterol response element (SRE) in the promoter area of the LDLR and HMG-CoA reductase genes, increasing their transcription. As the cholesterol content of the hepatocyte increases, the SREBP/SCAP complex is not incorporated into the ER, SREBP cannot reach the Golgi, and the NH2 terminal domain of SREBP cannot be released from the membrane for transport into the nucleus, and the transcription of the LDLR and HMG-CoA reductase genes decreases (12,14) (see also Fig. 8.3).
FIGURE 1.3 Schema of the LDLR pathway. LDLR are synthesized in the endoplasmic reticulum from which they enter the Golgi for glycosylation and then appear on the cell surface. The LDL aggregate in coated pits, bind the apoB-100 ligand on LDL, and the complex is internalized forming vesicles and endosomes. With the acidification of the endosomes, the LDL are targeted to the lysosomes wherein the apoB-100 moiety is degraded and the cholesteryl ester is hydrolyzed into free cholesterol and free fatty acids. The released cholesterol downregulates both the LDLR and HMG-CoA reductase genes. LDL receptors are recycled from the endosome back to the cell surface for reuse. ApoB-100, apolipoprotein B-100; LDL, low-density lipoproteins; HMG-CoA, hydroxymethylglutaryl-CoA. (Reprinted from Brown and Goldstein (12) with permission.)
When plasma low-density lipoprotein cholesterol (LDLC) exceeds 100 mg/dL, the capacity to process LDL through the LDLR pathway is exceeded. Increased numbers of LDL-particles cross the endothelial barrier, LDL are trapped in the vascular wall by proteoglycans, and then modified by either oxidation or glycation. Such modified LDL bind to scavenger receptor class A (SR-A1) and to CD36, a member of the scavenger receptor B family (Fig. 1.1), and enter cells such as macrophages by a low-affinity, LDL-receptor-independent mechanism. This alternate pathway is not subject to feedback inhibition of LDLR synthesis by LDL-derived cholesterol. Thus, LDL continues to be taken up in an unregulated fashion, leading to excess deposition of FC and CE in macrophages (Fig. 1.1). Dyslipidemias that favor an increased uptake of LDL through the scavenger pathway promote the production of foam cells and the associated atherosclerosis and xanthomas.
Lipoprotein Metabolism of ApoA-I-containing Lipoproteins: Major Receptors and Transporters
HDL can be differentiated from the apoB-containing lipoproteins by their comparably higher density, smaller size, and the presence of apoA-I.
Synthesis of HDL
Nascent HDL. ApoA-1 is released as a lipid-free protein from the intestine and liver (Fig. 1.2). ApoA-1 interacts with the ATP-binding cassette transporter 1 (ABCA1) on the basolateral membranes of hepatocytes, enterocytes, and macrophages acquiring PL and FC to form a more stable nascent HDL particle (5) (Fig. 1.2) (see also Chapter 9).
Formation of Larger, Mature HDL Particle
The transition of HDL particles from the disc-shaped nascent HDL to the spherical “mature” HDL requires the esterification of cholesterol to create a hydrophobic core. LCAT (Table 1.3) is associated with HDL in plasma and catalyzes the transfer of FFA from the PL, lecithin, to FC, forming CE. ApoA-I is a cofactor for this reaction (Fig. 1.2). CE formed by this reaction constitute the neutral lipid core of mature spherical HDL3; further activity of LCAT provides additional CE for the core of HDL3, forming the larger HDL2 particles (Fig. 1.2). Subsequent addition of cellular cholesterol to the HDL particle occurs in a number of tissues through the action of another ATP-binding cassette transporter 1 (ABCG1) and scavenger receptor class BI (SR-BI), molecules that prefer larger HDL as acceptors (Fig. 1.2).
In addition to acquiring lipids from liver and intestine, HDL also acquires lipids from chylomicrons and VLDL in the course of hydrolysis of TG by LPL. During this process, apoA-I is transferred from chylomicrons to HDL, and apoC-II and apoE on HDL are transferred to the TG-rich lipoproteins. The TG-rich lipoproteins shed excess PL and cholesterol that are transported to HDL by PLTP (5) (see also chapter 9).
Transfer of Lipid between HDL and the ApoB-containing Lipoproteins
CE are transferred from the core of the spherical HDL to TG-rich lipoproteins, a reaction promoted by CETP, which exchanges CE from HDL for TG from TG-rich lipoproteins (15) (Fig. 1.2). Thus, this process depletes CE from HDL and enriches TG in HDL and has important implications for HDL metabolism (5). For example, if the TG in HDL are hydrolyzed by HL, a smaller HDL particle is produced that is more avidly removed from plasma by cubulin in the kidney. PLTP is structurally similar to CETP and mediates the transfer of unsaturated fatty acids on PL from the apoB-containing lipoproteins to HDL, contributing to the acquisition of PL by HDL.
Reverse Cholesterol Transport
The CE on the spherical HDL can be transported back to liver by two mechanisms. CE are transferred from HDL to the apoB-containing lipoproteins by CETP, from which they are taken up by LDLR (Fig. 1.2). CE may also be delivered directly to the liver through SR-BI, also called the “HDL receptor.” These reactions are part of a process called reverse cholesterol transport. Once the CE is delivered to the liver and hydrolyzed into FC and FFA, the FC can be excreted directly into bile, or converted into bile acids by 7α-hydroxylase. Both these pathways result in delivery of sterol from peripheral tissues and plasma into intestinal cells, promoting the excretion of sterols into the stool. Reverse cholesterol transport has been postulated to explain, at least in part, the protective effect that HDL and apoA-I have against the development of atherosclerosis. Conversely, factors that impede this process, such as a dysfunctional HDL, appear to promote atherosclerosis (see also Chapter 9).
DISTRIBUTIONS OF LIPIDS, APOLIPOPROTEINS, AND LIPOPROTEINS
The plasma levels of lipids, apolipoproteins, and lipoproteins are distributed over a wide range of values (Tables 1.4, 1.5 and 1.6). This is not unexpected given the complexity of the major pathways for lipoprotein metabolism (Figs. 1.1 and 1.2), the multiple genes and proteins involved, and the influence of other factors, such as diet, weight, gender, and age. The genetic determinants of the plasma lipoprotein distributions have been elegantly reviewed by Robert Hegele (16). The distributions presented here are derived from Framingham, where all the measurements were performed in the same subjects and by the same laboratory methods (17,18,19,20). These distributions are for adults. Those for children are considered in Chapter 12. The extremes of these distributions are often used to define the quantitative aspects of the dyslipidemias associated with metabolic disorders of lipoprotein metabolism.
TABLE 1.4 ESTIMATED PERCENTILES OF LDL-C, NON-HDL-C, ApoB, AND LDL-P
Percentiles
5th
10th
25th
50th
75th
90th
95th
mg/dL
LDL-C
80
90
110
130
160
180
200
Non-HDL-C
110
120
140
160
190
210
230
ApoB
60
70
80
100
120
140
150
nmol/L
LDL-P
800
900
1,100
1,400
1,800
2,000
2,100
Estimates were derived from the Framingham Heart Study (17,18,19). Data for men and women were combined.
LDL-C, low-density lipoprotein cholesterol; Non-HDL-C, total cholesterol minus high-density lipoprotein cholesterol; ApoB, apolipoprotein B; LDL-P, total number of low-density lipoprotein particles.
TABLE 1.5 ESTIMATED PERCENTILES OF HDL-C AND ApoA-I
Percentiles
Sex
5th
10th
25th
50th
75th
90th
95th
mg/dL
Men
HDL-C
28
31
37
43
51
61
67
Women
HDL-C
35
39
46
55
66
77
84
Men
ApoA-I
92
99
114
130
153
178
196
Women
ApoA-I
107
116
132
154
181
206
224
Estimates were derived from the Framingham Heart Study (20). Data are presented separately for men and women.
TABLE 1.6 ESTIMATED PERCENTILES OF PLASMA TRIGLYCERIDES
Percentiles
Sex
5th
10th
25th
50th
75th
90th
95th
mg/dL
Men
46
55
75
112
173
252
324
Women
41
46
59
83
124
182
226
Estimates were derived from the Framingham Offspring Study (17). Data are presented separately for males and females.
GENETIC BASIS AND PHENOTYPIC PRESENTATION OF METABOLIC DISORDERS OF DYSLIPIDEMIA
The major biochemical complication of inherited dyslipidemias is the often profound disruption that occurs in either the quantity or quality, or both, of the plasma lipoproteins as the result of the expression of these genetic defects. The major complication of the inherited dyslipidemias is premature CVD. Atherosclerosis is the underlying, often silent process that results from dyslipidemias (see also Chapters 3 and 4). If other risk factors for CVD are also present, the atherosclerotic process is accelerated.
Another manifestation of the inherited dyslipidemias is the deposition of lipid mostly in skin, tendons, and the eyes. The composition of the lipid deposition in xanthomas reflects the underlying dyslipidemia. Patients with profound hypertriglyceridemia may develop eruptive xanthomas that are primarily composed of TG. Those with significant hypercholesterolemia manifest tendon xanthomas usually in the extensor tendons of the hands, the Achilles tendons, the tibial tuberosities, and elbows. CE are the major lipids found in tendon xanthomas. In sitosterolemia, approximately 15% of the esterified sterol in tendon xanthomas is plant sterol, reflecting the lipid composition of the LDL in this condition. Sterol esters can also be deposited prematurely in the cornea of the eyes (corneal arcus) and in the skin surrounding the eyes (xanthelasma). When xanthomas result from profound hypercholesterolemia, as occurs in children who have inherited a double dose of a mutant allele affecting LDLR activity (see the following text), sterol esters are deposited in the skin as somewhat raised yellow lesions (planar xanthomas), which are usually found between the webbing of fingers and toes, over the hands, in popliteal fossae, and over the buttocks.
Patients with profound hypertriglyceridemia can develop pancreatitis, a life-threatening condition in which the cells of the pancreas become inflamed and dysfunctional (see also Chapter 7). This has been attributed to the possibility of pancreatic lipase hydrolyzing the excess TG around the pancreas, with the FFA causing inflammation.
Only gold members can continue reading. Log In or Register to continue
Sep 7, 2016 | Posted by drzezo in ENDOCRINOLOGY | Comments Off on Lipid, Apolipoprotein, and Lipoprotein Metabolism: Implications for the Diagnosis and Treatment of Dyslipidemia
 Disorders of Hypertriglyceridemia
Disorders of Hypertriglyceridemia
 Dyslipidemia and Atherosclerosis in Women
Dyslipidemia and Atherosclerosis in Women
 Lipid, Apolipoprotein, and Lipoprotein Metabolism: Implications for the Diagnosis and Treatment of Dyslipidemia
Lipid, Apolipoprotein, and Lipoprotein Metabolism: Implications for the Diagnosis and Treatment of Dyslipidemia
 Disorders of Hypertriglyceridemia
Disorders of Hypertriglyceridemia
 Dyslipidemia in the Elderly: Special Insights to Inform the Management Strategy
Dyslipidemia in the Elderly: Special Insights to Inform the Management Strategy
 Trans Fatty Acids, Dyslipidemia, and Cardiovascular Disease
Trans Fatty Acids, Dyslipidemia, and Cardiovascular Disease



