Disease
Clinical characteristic
Histologic features
CD1a
CD14
CD68
CD163
CD207
S100
Factor XIIIa
Fascin
HLA-DR
Others
Langerhans cell histiocytosis
See text
+
−
+/−
−
+
+
−
−
+
Birbeck granules
Juvenile xanthogranuloma
Cutaneous type (75%)
Mainly <2 years old, multiple nodular small papules
−
+
+
+
−
−
+
+
−
Touton giant cells (cutaneous type)
Soft tissue type (15%)
Mainly <2 years old, soft tissue mass lesions in subcutaneous fat or muscle
Systemic type (10%)
Mainly <6 months old, mass lesions in visceral organs, subcutis, central nervous system, bone, liver, spleen, lung,and others
Erdheim-Chester disease
Mainly >40 years old
Symmetrical sclerotic lesions in the long bones
Nonosseous lesions—ureteral stricture associated with retroperitoneal fibrosis, xanthomatous skin lesions, central diabetes insipidus, and pulmonary fibrosis (50%)
−
+
+
+
−
−
+
+
−
Foamy histiocytes
Rosai-Dorfman disease
Children and young adults
Painless bilateral cervical lymphadenopathy (80%)
Extranodal lesions—osteolytic bone lesions, maculopapular skin rash, orbital masses, and others (50%)
−
+
+
+
−
+
−
+
+
Emperipolesis
Langerhans cell sarcoma
Any age
Multiple organ involvements—lymph nodes, liver, spleen, bone, lung, and others
+
+
+
−
+
+
−
+/−
+
Nuclear pleomorphism and mitoses
11.4 Pathogenesis
It is debatable whether LCH is a neoplasm or inflammation. Recently, LCH was defined as an inflammatory myeloid neoplasia [14]. Proliferation of LCH cells is neoplastic, although some clinical presentations are due to inflammation induced by infiltrating inflammatory cells, as well as LCH cells.
11.4.1 LCH and Neoplasia
11.4.1.1 Somatic Mutation of LCH Cells
LCH cells are monoclonal [15, 16] and sometimes show chromosomal deletion or gain [17]. However, recurrent genomic alteration was not detected for a long time due to the low percentage of LCH cells in the lesion [18]. However, a report published in 2010 revealed that LCH cells harbor an oncogenic mutation of the BRAF gene (V600E), which plays a role in the mitogen-activated protein kinase (MAPK) signaling pathway; it also showed that extracellular signal-regulated kinase (ERK), a molecule downstream of BRAF in this pathway, is phosphorylated constitutively in the presence or absence of this mutation [19]. This mutation is detected in about half of LCH patients, regardless of disease type [20]. The clinical significance of the BRAF V600E mutation is unclear, although it was reported recently that BRAF V600E mutation-positive patients may experience a higher rate of reactivation than mutation-negative patients [21]. More recently, oncogenic mutation of other genes within the MAPK signaling pathway, including ERBB3, ARAF, and MAP2K1, were identified in LCH cells [22–24] (Fig. 11.1). These MAPK signaling pathway-related mutations are mutually exclusive. Recurrent loss-of-function mutations in MAP3K1 (which encodes MEKK1) in the c-Jun N-terminal kinase pathway were also identified [25]. These mutations of MAP3K1 are not exclusive to the BRAF V600E mutation, and the mechanisms by which these variants promote neoplastic growth remain unknown.
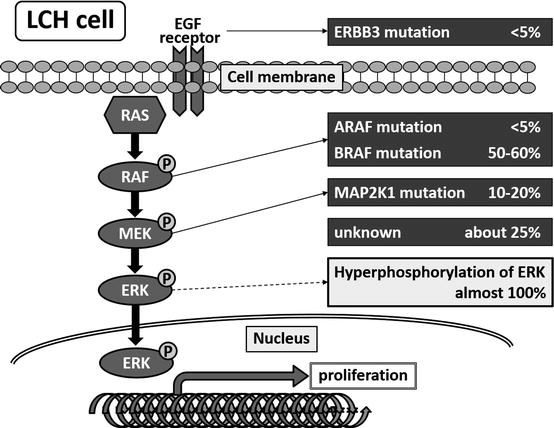
Fig. 11.1
Somatic mutations in the mitogen-activated protein kinase signaling pathway in LCH cells
11.4.1.2 Characteristic of Neoplastic LCH Cells
Gene expression analysis of purified LCH cells revealed that the LCH cell of origin is not a skin Langerhans cell but a myeloid DC [26]. Compared with normal Langerhans cells, LCH cells are more proliferative and have a lower antigen-presenting capacity. During maturation, LCH cells arrests in an activated state, possibly due to the action of transforming growth factor-β and interleukin (IL)-10 [27]. LCH cells are the only DCs that co-express Notch1 and its ligands Jagged-1 and -2 [28]. The JAG-mediated Notch signaling pathway may also play an important role in maintaining LCH cells in an immature state. Leukemic cells and LCH cells in patients who develop T cell acute lymphoblastic leukemia before LCH harbor the same activating Notch1 mutation and T cell receptor rearrangements [29, 30], further supporting the possibility that Notch1 may contribute to LCH pathogenesis. This is also interesting when considering the origin of LCH cells.
11.4.1.3 Detection of BRAF V600E-Positive Cells in Peripheral Blood and Bone Marrow
BRAF V600E mutations are detected in bone marrow hematopoietic progenitors, peripheral monocytes, and myeloid DCs in active high-risk (RO-positive MS) LCH patients harboring a BRAF V600E mutation. Conversely, the mutation is restricted to lesional LCH cells in low-risk patients [21].
11.4.1.4 Animal Model of LCH Generated by Forced BRAF V600E Expression in DCs
Forced expression of BRAF V600E in DCs is sufficient to drive LCH-like disease in mice [21]. Forced expression of BRAF V600E in CD11c+ cells (DC progenitors) results in the formation of aggressive LCH lesions similar to those observed in humans with high-risk LCH, whereas BRAF V600E expression in langerin-positive cells (differentiated DCs) results in disease that resembles low-risk LCH without detectable mutations in circulating myeloid cells. These data suggest that the extent of LCH may be determined by whether ERK-activating mutations arise at any stage of DC differentiation.
11.4.2 LCH and Inflammation
11.4.2.1 Interaction Between LCH Cells and Inflammatory Cells That Infiltrate LCH Lesions
LCH lesions contain less than 10% (median value) LCH cells [21]; however, they contain many inflammatory cells, including eosinophils, T lymphocytes, plasma cells, macrophages, osteoclast (OC)-like multinucleated giant cells (MGCs), and neutrophils. Because LCH cells show immature DC characteristics, they may promote the expansion of regulatory T cells, which in turn results in the failure of the host immune system to eliminate LCH cells [31].
LCH cells express the immature DC marker CCR6 and secrete its ligand CCL20/MIP-3α, along with CCL5/RANTES (a CCR1/3/5 ligand) and CXCL11/I-TAC (a CXCR3 ligand) [32]. Interactions between these chemokine receptors and their ligands may play a role in the hematogenous migration of not only LCH cells but also of inflammatory cells into the lesions. These LCH cells and infiltrating cells in turn stimulate each other to produce cytokines such as GM-CSF, interferon (IFN)-γ, tumor necrosis factor (TNF)-α, IL-1α, IL-2, IL-3, IL-4, IL-5, IL-7, and IL-10 [33]. Moreover, LCH cells in lesions express high levels of CD40, whereas T cells express CD154 (the CD40 ligand): interactions between CD40 and CD154 are essential for activation of both antigen-presenting cells and T cells [34]. A recent report shows that LCH cells and lesional T cells express high levels of the pleiotropic cytokine osteopontin (OPN) [26], which promotes the generation of T helper (Th)1 and Th17 cells, recruits histiocytes/monocytes, and activates osteoclasts [35].
We found that the serum levels of lesional inflammatory cytokines/chemokines and DC/macrophage-activating factors such as IL-18, M-CSF, and CCL2 are markers of disease dissemination and severity [36]. Marked increases in serum IL-18 and soluble tumor necrosis factor receptors are reported in patients at risk for developing hemophagocytic lymphohistiocytosis [37]. In addition, we found that patients with MS LCH have much higher serum OPN levels than patients with SS LCH [38].
11.4.2.2 Osteoclastogenesis in LCH Lesions
In LCH, OC-like MGCs are present not only in the bone but also in skin and lymph node lesions, and enzymes derived from OC-like MGCs may play a major role in chronic tissue destruction [39]. Both LCH cells and T cells in lesions produce the OC-inducing cytokines, receptor activator for nuclear factor kB ligand (RANKL) and macrophage colony-stimulating factor (M-CSF) [39]. We found that patients with SS LCH and multiple bone lesions had significantly higher ratios of soluble RANKL and osteoprotegerin (OPG), a decoy receptor of RANKL, than patients with MS LCH; moreover, the soluble RANKL/OPG ratios correlated positively with the number of bone lesions [40].
Courey et al. showed that patients with active LCH have high serum levels of IL-17A, which is mainly produced by LCH cells [41]. In vitro, serum IL-17A causes fusion of immature DCs, which results in formation of OC-like MGCs [41]. This IL-17A-dependent fusion activity correlates with LCH activity. An IL-17A autocrine model of LCH was suggested. We also found that patients with LCH have higher serum levels of IL-17A than controls, although the serum IL-17A levels of patients with MS LCH and SS LCH did not differ significantly. However, we also observed that LCH cells in MS disease expressed higher levels of IL-17A receptor (IL-17RA) than those in SS disease and that IL-17RA expression levels help distinguish between LCH subclasses [42]. In addition, we found that the cleaved form of OPN plays a critical role in driving immature DC differentiation into OC-like MGCs in an autocrine and/or paracrine fashion [43].
11.4.2.3 Triggers of LCH
Merkel cell polyomavirus (MCPyV, a common dermotropic virus) sequences are detected in not only skin but also in bone and soft tissue lesions of LCH patients; also, the amount of MCPyV DNA in peripheral blood cells is increased in patients with high-risk (RO-positive MS) LCH but not in patients with low-risk LCH [44]. MCPyV infection may induce LCH cells with an oncogenic mutation to secrete IL-1; this leads to activation of LCH cells in an autocrine manner [45]. This suggests that MCPyV infection may trigger LCH.
Nicotine stimulation increases the production of both OPN and GM-CSF by alveolar macrophages. Bronchoalveolar lavage cells from patients with pulmonary LCH produce abundant amounts of OPN. Forced expression of OPN in rat lungs induces lesions similar to pulmonary LCH, with marked accumulation of Langerhans cells [46]. These findings suggest that increased OPN production upon stimulation by nicotine is responsible for pulmonary LCH.
11.5 Clinical Manifestations
Since LCH affects a number of different organs, clinical signs and symptoms can be extremely variable. In most pediatric patients with SS LCH, lesions occur in the bone, with either single-site or multifocal involvement; however, lesions can also occur in the skin, thymus, or lymph nodes [47]. The involved organ at diagnosis and the initial presentation of MS LCH are shown in Fig. 11.2. In patients with MS LCH, the organs involved are mainly the bone and skin, followed by the hematopoietic system, lymph nodes, liver, spleen, lung, and thymus. The most common initial manifestations are development of a skin rash, a soft tissue mass, fever, lymphadenopathy, hepatosplenomegaly, polyposia/polyuria, and bone pain. There are no specific laboratory markers for LCH. In many cases, LCH presents with nonspecific inflammatory signs that arise from chronic inflammation.
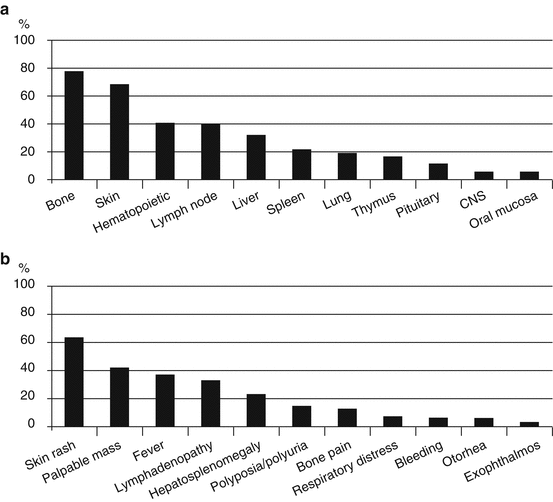
Fig. 11.2
(a) Involved organ and (b) Initial presentation of patients with multi-system Langerhans cell histiocytosis at the time of diagnosis (n = 206; JLSG-96/02 study)
11.5.1 Bone Lesions
Bone is involved in about 80% of all LCH cases [48]. The skull is the most common location, followed by the spine, extremities, pelvic bone, and ribs [49]. X-rays typically show osteolytic “punched out” lesions with sharp margins. Fluorodeoxyglucose-positron emission tomography [50] and whole-body magnetic resonance imaging (MRI) [51] are also useful for detecting bone lesions and for evaluating their response to treatment. Bone lesions may be asymptomatic or accompanied by pain and soft tissue swelling. When LCH is localized solely to the bone, the clinical course is generally benign and sometimes resolves spontaneously over a period of months to years. In patients with RO-positive MS disease, lack of bone lesions at the time of diagnosis is associated with a poorer outcome [52]. Bone lesions can result in critical or irreversible symptoms, such as visual loss or exophthalmos (due to orbital involvement), conductive hearing loss (due to mastoid antrum lesions), loss of teeth (due to jaw disease), and spinal paralysis (due to vertebral lesions).
11.5.2 Skin Lesions
Skin involvement is seen in approximately half of all patients and is particularly common in infants. These patients present with various lesions, including erythema, papules, nodules, petechiae, vesicles, crusted plaques, and seborrhea-like eruptions. Ulcerative lesions in the genital or inguinal region may also be present [53]. It is essential that LCH patients with skin lesions undergo evaluation of other visceral organs because most of these patients have MS disease. Patients with skin involvement whom are aged older than 18 months are more likely to have lesions in organs other than the skin [54]. In neonatal infants, isolated skin lesions can regress spontaneously over weeks to months [55, 56]. However, some infants with isolated skin lesions at the time of diagnosis can develop MS disease, followed by a fatal clinical course [57].
11.5.3 Risk Organs (Hematopoietic System, Liver, and Spleen)
Because disease involving the hematopoietic system, liver, or spleen is associated with a higher risk of death, these organs are defined as “risk organs”. Pulmonary involvement is no longer considered an independent prognostic variable [58, 59]. The recent definition of risk-organ involvement is shown in Table 11.2 [60].
Hematopoietic involvement (with or without bone marrow involvement) |
Both of the following should be present: |
– Hemoglobin less than 10 g/dL (not due to other causes, e.g., iron deficiency) |
– Thrombocytopenia, with a platelet count less than 100,000/mm3 |
Liver involvement (the patient can show a combination of these symptoms): |
– Enlargement >3 cm below the costal margin at the midclavicular line, confirmed by ultrasound |
– Dysfunction documented by hyperbilirubinemia >3 times normal hypoalbuminemia (<30 g/dL); γ GT increased >2 times normal; ALT/AST >3 times normal; ascites or edema |
– Intrahepatic nodular mass |
Spleen involvement |
– Enlargement >3 cm below the costal margin at the midclavicular line, confirmed by ultrasound |
Hematopoietic system involvement is defined by cytopenia, either with or without bone marrow infiltration of LCH cells, and is seen exclusively in MS disease. In severe cases, serious anemia and thrombocytopenia may develop and are often associated with a secondary hemophagocytic syndrome, followed by a fatal course. In particular, anemia with thrombocytopenia, with or without leukopenia and hypoalbuminemia, is associated with a worse prognosis in children with MS LCH [61].
Liver and/or spleen involvement is characterized by organomegaly and/or liver dysfunction and is observed exclusively in MS disease. Histological examination of the liver reveals a portal infiltrate, which can lead to destruction of bile ducts and periportal fibrosis (sclerosing cholangitis), culminating in biliary cirrhosis with portal hypertension and, ultimately, secondary hypersplenism, which can cause cytopenias. A liver transplant is the treatment of choice for patients with sclerosing cholangitis due to LCH [62].
11.5.4 Lung Lesions
In children, pulmonary involvement is usually associated with MS disease. Lung pathology is associated with cough, dyspnea, pleural effusion, and recurring pneumothorax. High resolution-computed tomography may reveal reticular or micronodular opacity, as well as large nodules and honeycombing. KL-6 may be a useful marker for pulmonary involvement [63]. Severe pulmonary hypertension is a common feature in patients with end-stage pulmonary LCH. Lung transplantation is a therapeutic option in this setting [64].
11.5.5 CNS Lesions
LCH may involve the CNS, presenting as space-occupying mass lesions, hypothalamic-pituitary disease, or neurodegenerative disease.
11.5.5.1 Mass Lesions
11.5.5.2 Hypothalamic-Pituitary Disease
Infiltration and dysfunction of the pituitary gland and/or adjacent hypothalamus is observed in 25% of all patients with LCH and in 50% of patients with MS disease [65]. The most frequent manifestation is CDI, which usually follows (but sometimes precedes or co-occurs with) other symptoms and signs of the disease. The cumulative incidence of CDI continues to increase, even 10 years after a diagnosis of LCH [67]. CDI occurs more often in patients with craniofacial bone lesions (with the exception of the vault), ear, eye, and/or oral lesions [68]; thus these lesions are known as CNS-risk lesions. Patients with MS disease that includes bone lesions are more likely to have CNS-risk lesions than patients with bone lesions alone [49]. LCH involvement of the pituitary gland can be shown by MRI, namely, pituitary stalk thickening and loss of the physiological high-intensity signal in the posterior pituitary lobe on T1-weighted images. Appropriate systemic chemotherapy may effectively inhibit the development of CDI [67, 68]. Systemic chemotherapy rarely cures CDI, but LCH patients with newly diagnosed CDI should be treated with systemic chemotherapy to prevent anterior pituitary hormone loss and neurodegenerative disease. About half of patients with CDI develop anterior pituitary hormone deficiencies and/or ND-CNS disease during follow-up, usually after a disease course of several years [69]. Patients with hypothalamic-pituitary lesions may present with non-endocrine hypothalamic dysfunction, such as eating disorder, thirst, fatigue, autonomic disturbance, temperature instability, memory deficit, and disturbed consciousness [70]. Hypothalamic lesions are observed on MRI as gadolinium-enhanced masses [71].
11.5.5.3 Neurodegenerative CNS Disease
ND-CNS disease may develop during the years after disease onset, often when the disease is considered quiescent [72, 73]. Three years after a diagnosis of LCH, more than half of patients with CNS-risk lesions and/or CDI demonstrate abnormities on MRI (radiological ND-CNS disease), including bilateral symmetrical lesions in the cerebellar white matter and/or basal ganglia [74]. These brain MRI abnormalities gradually deteriorate and are irreversible. One quarter of patients with brain MRI abnormalities present with neurological symptoms (neurological ND-CNS disease), which includes ataxia, tremor, dysarthria, dysphagia, and hyperreflexia, within several years, which is followed by further deterioration. Patients with neurological ND-CNS disease eventually become bedridden. Histologically, ND-CNS disease is characterized by the presence of a CD8-positive T cell infiltrate, activation of microglia, gliosis, and neuronal and axonal destruction with secondary demyelination, as observed in autoimmune encephalitis. CD1a-positive LCH cells may be absent [75]. Currently, there is no established therapy for ND-CNS disease, although promising evidence suggests that intravenous immunoglobulin [76, 77] can prevent progression. Although first-line therapies for LCH that include cytarabine (Ara-C) cannot prevent development of ND-LCH completely [78], treatment with Ara-C might be effective for ND-CNS disease [79].
11.5.6 Other Lesions
The most commonly affected lymph nodes are the cervical nodes, followed by axillary and inguinal nodes [80]. Rarely, lymph nodes can become massive and cause upper airway obstruction. Thymic involvement mainly occurs in young children and is diagnosed when a CT indicates thymus enlargement and calcification. Patients with thymic involvement sometimes have profound T lymphocytopenia and (albeit rarely) suffer superior vena cava syndrome or asphyxiation [81]. Infiltration of the oral mucosa may appear as ulceration or swelling of the gingiva [82]. Infiltration of the gastrointestinal tract rarely occurs, but can cause vomiting, abdominal pain, constipation, intractable diarrhea with blood and/or mucus [83], and malabsorption of nutrients [84]. Occasionally, the thyroid gland [85], pancreas, and kidneys [86] are involved.
11.6 New Clinical Markers of LCH
In patients with various solid tumors, cell-free DNA (cfDNA) in the peripheral blood contains cancer-derived genomic DNA and can be tested for using a noninvasive diagnostic procedure called a “liquid biopsy.” cfDNA BRAF V600E in plasma may serve as a noninvasive biomarker of treatment response in high-risk LCH patients [87, 88].
Acute-phase serum levels of inter-alpha-trypsin inhibitor heavy chain 4 (ITIH4), whose production is up-regulated by IL-6, may represent disease activity or extent of LCH [89].
11.7 Treatment and Outcome
The clinical course of LCH varies widely depending on the extent of organ involvement. The mortality rate of patients with SS disease, or RO-negative MS disease, is less than 1%. However, children with RO-positive MS disease have a worse prognosis: these children often have fatal outcomes despite intensive treatment and mortality rates are 10–50% [90]. Treatment of LCH should be planned according to the clinical presentation and the extent of organ involvement. Etoposide (VP-16) is no longer considered a reasonable first-line therapeutic agent because it is no more efficacious than vinblastine [91] and causes therapy-related acute myeloid leukemia (t-AML) [92]. Radiation is rarely used in children because of the increased risk of secondary malignancies, particularly brain tumors, in irradiated areas [93].
11.7.1 Single-System LCH
In SS LCH, the main aim of treatment is to reduce the symptoms and the chances of permanent sequelae. In patients with localized, unifocal bone disease, LCH may resolve spontaneously. In the case of a single bone lesion in a region other than the craniofacial area, or in those with no symptoms, a wait-and-see approach or diagnostic curettage is the standard method of care. Local injection of corticosteroid may be used for these bone lesions [94]. Patients with CNS-risk lesions with compressing soft tissue extension of bone lesions or multifocal bone disease should receive systemic chemotherapy with vinca alkaloids and corticosteroids for more than 6 months to prevent late sequelae such as CDI and nerve compression [95]. A nationwide survey in Japan suggests that appropriate chemotherapy for patients with a single bone lesion results in an excellent prognosis [47]. Radical resection of large bone lesions is discouraged as this often results in disfigurement. In cases of skin involvement only, the best option is to take a wait-and-see approach, with careful attention paid to progression to MS disease. Alternatively, patients can be treated with topical corticosteroids.
Related posts:
 Inherited Bone Marrow Failure Syndrome, TAM
Inherited Bone Marrow Failure Syndrome, TAM
 Childhood Aplastic Anemia
Childhood Aplastic Anemia
 Hemophagocytic Lymphohistiocytosis, Secondary
Hemophagocytic Lymphohistiocytosis, Secondary
 Primary Hemophagocytic Lymphohistiocytosis
Primary Hemophagocytic Lymphohistiocytosis
 Myelodysplastic Syndrome (MDS) and Juvenile Myelomonocytic Leukemia (JMML)
Myelodysplastic Syndrome (MDS) and Juvenile Myelomonocytic Leukemia (JMML)
 Hematopoietic Stem Cells: The Basis of Normal and Malignant Hematopoiesis
Hematopoietic Stem Cells: The Basis of Normal and Malignant Hematopoiesis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


