Chapter 74 Lactic Acidosis and Other Mitochondrial Disorders Associated with HIV Therapy
INTRODUCTION
Nucleoside analog reverse transcriptase inhibitors (NRTI) were the first class of drugs to demonstrate potent efficacy against HIV infection, being introduced into clinical practice in 1987 (with the advent of zidovudine monotherapy) within a short period of the widespread recognition of AIDS and of HIV as its underlying cause. However, the ongoing use of NRTI therapy into the second decade of HIV therapy highlights the need for a comprehensive understanding of NRTI-induced drug toxicities,1–3 as HIV infection becomes a more manageable disease with greatly reduced morbidity and mortality.4,5 This chapter will review current knowledge of the role of mitochondrial toxicity in the pathogenesis of clinical toxicity syndromes associated with NRTI drug therapy, including lactic acidosis as well as a number of tissue-specific treatment complications (e.g., lipoatrophy, neuropathy).
The risk of developing severe toxicities such as lactic acidosis appears to have declined significantly in recent years.6 However, it is also apparent that certain long-term HIV drug toxicities provide an ongoing burden of disease, so that established clinical syndromes may not improve substantially even years after the removal of the causative NRTI drug. In this context, there is an ongoing need not only to prevent NRTI complications wherever possible, but also to develop effective treatment strategies for those affected by these treatment complications.
MODE OF ACTION OF NRTI DRUGS
The antiretroviral efficacy of NRTI drugs is conferred by their ability to inhibit HIV reverse transcriptase, the viral RNA-dependent DNA polymerase that allows the virus to create a DNA sequence based on its own RNA template (Fig. 74-1). Following intracellular phosphorylation, NRTI drugs compete with naturally occurring nucleotides for utilization by HIV reverse transcriptase. Incorporation of the “false” NRTI substrate then causes DNA chain termination, as these nucleotides lack a hydroxyl group that is critical for chain elongation. The effectiveness of NRTI drugs as inhibitors of HIV replication is therefore determined primarily by their ability to inhibit HIV reverse transcriptase. However, the efficiency with which NRTI drugs are delivered to the appropriate cell population and intracellular compartment–determined by factors such as oral bioavailability, cellular transport mechanisms, and the efficiency of cellular kinases responsible for activating NRTI drugs to their triphosphate form–is also an important factor in determining drug efficacy.
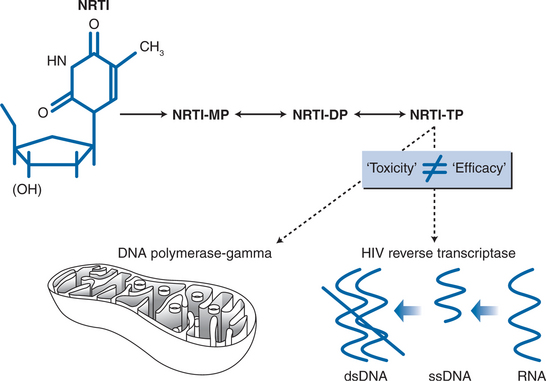
POTENTIAL MECHANISMS OFNRTI-MEDIATED MITOCHONDRIAL TOXICITY
The study of NRTI-associated toxicity syndromes has also been possible due to information available on the pharmacology of these drugs,1–3 and particularly by their capacity to inhibit a specific host DNA polymerase (mitochondrial DNA polymerase-gamma) through a mechanism that is similar to its therapeutic activity as an HIV reverse transcriptase inhibitor. However, it is worth stating at the outset that the toxic and therapeutic profiles of individual NRTI drugs are not correlated, so that an increased efficacy is not necessarily accompanied by increased toxicity.7 Indeed, over the past 15 years there has been an increasing trend toward using NRTI drugs that combine high levels of antiviral efficacy with benign toxicity profiles. This trend toward reduced toxicity may also reflect the avoidance of NRTI combinations that have synergistic toxicity profiles, as may be seen in the case of NRTI-associated neuropathy where stavudine and didanosine each contribute to increased risk. This will be discussed further.
Mitochondria are the energy powerhouses of the cell, and are unique among intracellular organelles in containing their own genome that is extrachromosomal and replicates independently of nuclear DNA. Hence, mitochondrial DNA can be synthesized in postmitotic and resting cells, utilizing a unique DNA polymerase (polymerase-gamma) whose evolutionary origins8 and enzymatic activities9 are distinctfrom the multiple DNA polymerases involved in nuclear DNA synthesis. Importantly, DNA polymerase-gamma is also unique among the human polymerases in that it is unable to discriminate between naturally occurring nucleotides and nucleoside analogue drugs.8 Hence, NRTI drugs are capable of inhibiting cellular mitochondrial DNA synthesis via their effects on DNA polymerase-gamma (Fig. 74-1). The proposal that NRTI-induced polymerase-gamma inhibition represents a common pathway for tissue-specific adverse effects of this drug class has been reviewed extensively by Lewis and Dalakas,10 Brinkman,11 and more recently by Kakuda4 and White.5 The basic premise of the “pol-gamma” hypothesis is that NRTI-induced inhibition of this DNA polymerase leads to cellular mitochondrial DNA depletion (i.e., reduced numbers of mitochondrial DNA copies per cell). Cellular, and ultimately organ toxicity is the consequence of loss of mitochondrial bioenergetic function, when mitochondrial DNA levels have fallen beyond a critical level at which the production of the 13 mitochondrial DNA-encoded protein subunits of the mitochondrial respiratory chain is insufficient to meet energy requirements (see Fig. 74-2).
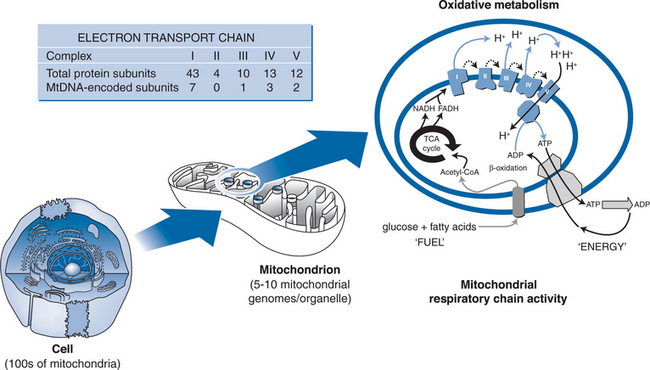
At the cellular level, there is an increasing understanding that while the ability of a specific NRTI to induce mitochondrial DNA depletion is primarily determined by its relative affinity for polymerase-gamma, the relevance of this effect to the development of tissue toxicity is modulated by other factors, such as that drug’s ability to access the mitochondrial compartment within the cell cytosol in an activated form. This is highlighted by the recent discovery of a mitochondrial inner membrane transporter that is able to facilitate the entry of active NRTI diphosphate and triphosphate derivatives into mitochondria.12
In addition to the mechanism described above, zidovudine may exert at least some of its mitochondrial toxicity effects through “non-pol-gamma” mechanisms, with a body of research evidence indicating that zidovudine has direct effects on critical components of the mitochondrial membrane involved in cellular energy metabolism (e.g., the ADP/ATP transporter that exports mitochondrial-derived ATP to the cytosol).13,14 In this context, it is also interesting to note that the mitochondrial ADP/ATP transporter also appearsto be a target for selected HIV protease inhibitors (PIs) such as nelfinavir, which are thought to exert antiapoptoticeffects through mitochondrial membrane stabilization.15 However, this effect, now well-characterized in a number of in vitro and in vivo studies, is yet to find a widespread clinical application.
It is also important to recognize that inhibiting mitochondrial DNA polymerase-gamma may affect the fidelity of mitochondrial DNA synthesis–resulting in increased risk of mitochondrial DNA mutation–as well as causing mitochondrial DNA depletion.16,17 The clinical importance of this effect has not been established, although neurologic syndromes attributable to mitochondrial DNA mutations have been seen in association with NRTI therapy, including five published cases of Leber’s hereditary optic neuropathy (characterized by rapidly evolving bilateral optic atrophy and blindness).18–21 It is likely that these individuals were genetically susceptible to this condition, in which the development of the clinical phenotype generally requires a very high proportion of mutated mitochondrial DNA, and that NRTI therapy may have “unmasked” the syndrome. However, the development of an unusual neurologic syndrome in an NRTI-treated individual should alert the clinician to the possibility of a “mitochondrial disease”. Similarly, mitochondrial DNA mutation may be considered in rare cases in which mitochondrial dysfunction appears to be progressive despite cessation of NRTI therapy.
MITOCHONDRIAL TOXICITY AND PREGNANCY: FETAL AND MATERNAL CONSIDERATIONS
Before returning to more “typical” manifestations of mitochondrial toxicity associated with NRTI therapy, there has been concern that fetal exposure to NRTI therapy in utero may lead to the development of mitochondrial disease in infants, following the report of eight children with consistent clinical features.22 A comprehensive US epidemiologic study examining this issue (n = 1954, including 188 deaths at age <5 years, with evidence of mitochondrial dysfunction rated from “unlikely” (class 1) to “highly likely/proven” (class 4)) has not identified increased mortality attributable to mitochondrial disease among NRTI-exposed infants,23 and concluded that the benefits of perinatal zidovudine therapy are likely to outweigh the risks.23,24 However, the French investigators who initially described this association have gone on to demonstrate an 18-month incidence of “established” mitochondrial dysfunction (i.e., clinical syndrome supported by histopathology and evidence of mitochondrial respiratory chain defects) among ∼0.3% infants in a similarly large population-based study (7/2644 cases among NRTI-exposed infants, 0/1748 in unexposed controls).25 “Possible” mitochondrial dysfunction was identified in a further 14 NRTI-exposed infants. This study also identified a relatively coherent clinical syndrome, consisting of (1) neurological symptoms (developmental delay, seizures, behavioral disturbances); (2) cerebral magnetic resonance imaging (MRI) abnormalities (white matter and brainstem lesions)26; and (3) hyperlactatemia. Risk factor analysis including both “possible” and “established” cases suggested increased risk associated with combined NRTI therapy (15/21 cases; zidovudine + lamivudine, n = 14) compared with zidovudine monotherapy (6/21 cases) (RR = 2.5, 95% CI 1–6.5). Intravenous zidovudine therapy was utilized in 20/21 cases. However, duration of NRTI therapy was not found to be different among cases and controls, and mitochondrial DNA depletion was not found in affected tissues despite the presence of histopathological and biochemical evidence of mitochondrial toxicity. The authors stressed that these findings do not argue against the usefulness of NRTI therapy in pregnancy.
With respect to the risks of maternal mitochondrial toxicity associated with NRTI use in pregnancy, however, there is increasing evidence that stavudine therapy, particularly when combined with didanosine, is associated with risk of lactic acidosis in late pregnancy.27,28 This has prompted the manufacturers to issue product information in 2001 warning that the combination of stavudine and didanosine should be avoided during pregnancy “unless the potential benefit clearly outweighs the potential risks”.29
IN VITRO STUDIES: ESTABLISHING TOXICITY PROFILE FOR NRTI DRUGS
In vitro studies have provided important information regarding the potential of specific NRTI drugs to cause DNA polymerase-gamma inhibition, mitochondrial DNA depletion, and mitochondrial dysfunction.9,30–32 A consistent ranking of the potency of mitochondrial DNA synthesis inhibition has emerged from these studies, with decreasing potency associated with ddC (zalcitabine)  ddI (didanosine) > d4T (stavudine) > AZT (zidovudine). These studies also suggest that no significant mitochondrial polymerase-gamma inhibition occurs at pharmacological concentrations of lamivudine, abacavir, and tenofovir. One caveat to these findings is that NRTI-associated mitochondrial toxicity is tissue-specific, so that in vitro studies should ideally utilize human cell cultures that are relevant to the clinical toxicity profile of the drug.
ddI (didanosine) > d4T (stavudine) > AZT (zidovudine). These studies also suggest that no significant mitochondrial polymerase-gamma inhibition occurs at pharmacological concentrations of lamivudine, abacavir, and tenofovir. One caveat to these findings is that NRTI-associated mitochondrial toxicity is tissue-specific, so that in vitro studies should ideally utilize human cell cultures that are relevant to the clinical toxicity profile of the drug.
CLINICAL NRTI-INDUCED TOXICITY SYNDROMES
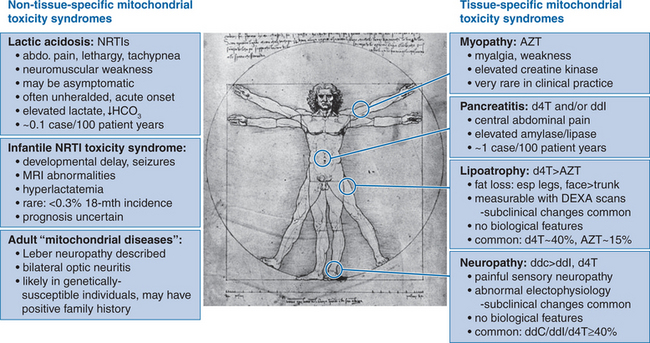
While the prototypic NRTI-associated toxicity syndrome is zidovudine myopathy, this syndrome is now rare in clinical practice,33,34 possibly reflecting either the use of lower zidovudine doses in the HAART era, or the involvement of uncontrolled HIV infection per se in the pathogenesis of this syndrome. Currently, the most concerning adverse effects involving NRTI therapy are lactic acidosis and other less serious disorders of lactate metabolism; the progressive subcutaneous fat wasting syndrome that is viewed as part of the lipodystrophy syndrome; neuropathy; and pancreatitis (Fig. 74-3).
LACTIC ACIDOSIS AND HYPERLACTATEMIA
Lactic acidosis is probably the most recognizable feature of mitochondrial dysfunction in clinical disease, in which loss of mitochondrial oxidative function leads to increased reliance on “anaerobic” metabolism and the inevitable accumulation of lactate (and thus, of acid). In the setting of NRTI therapy, there is now an appreciation of a spectrum of clinical phenotypes associated with elevated systemic lactate levels.35 At one end of this spectrum is a relatively common syndrome of mild, asymptomatic, nonprogressive hyperlactatemia (generally <2.5 mmol/L), which appears to represent a “compensated” homeostatic system in which elevated lactate production is balanced by effective mechanisms of lactate clearance. While the degree of hyperlactatemia appears to be greater in the presence of stavudine or didanosine therapy compared with zidovudine or abacavir, this syndrome appears to be benign irrespective of the choice of NRTI therapy.36–40
In contrast, lactic acidosis and hepatic steatosis represents a relatively uncommon (∼1–2 per 1000 person-years) but life-threatening clinical syndrome in which lactate homeostasis is completely “decompensated”, allowing the rapid, progressive accumulation of lactate and development of acidosis in affected patients. A critical aspect of lactic acidosis is its unpredictability, as it typically occurs in patients who have been on stable NRTI regimens for months or even years, and is not generally heralded by increased lactate levels before the development of the fulminant syndrome.36,41 Host risk factors have been identified for NRTI-associated lactic acidosis, including concurrent liver disease, female gender42 (reviewed in Chapter 42), and obesity. The association between female gender and risk of NRTI-induced lactic acidosis is a striking one, with a United States Food and Drug Administration (FDA) report of 60 cases finding that 83% occurred in women, including 85% of fatal cases.43 In relation to drug treatment, the majority of reported cases in recent years have involved stavudine-based HAART, although it is important to recognize that cases involving zidovudine therapy also occur (reviewed in Chapters 35 and 41). It is encouraging to note that a Swiss HIV cohort study has documented a declining incidence of both hyperlactatemia (lactate >2.4 mmol/L) and lactic acidosis (lactate >5 mmol/L) over time, with hyperlactatemia and lactic acidosis rates progressively falling by ∼60% and 85%, respectively, in the interval between 1999 and 2003.6 In this study, this pattern appeared to reflect reduced prescription of stavudine/didanosine dual NRTI therapy over the study period.
The cornerstone of management of this condition is early recognition of the clinical manifestations: abdominal symptoms including nausea, vomiting, anorexia, abdominal pain and distension, fatigue, with biochemical evidence of hepatocellular liver dysfunction and lactate level generally >5 mmol/L. These clinical and laboratory abnormalities should lead to prompt cessation of NRTI therapy. A more recently identified clinical syndrome accompanying lactic acidosis is progressive, severe neuromuscular weakness mimicking the Guillain–Barré syndrome.44 Initial reports by the FDA identified 25 cases, including seven fatal cases (notably, NRTI therapy was continued after the diagnosis was made in all but one of these individuals). A more recent retrospective analysis has identified 55 cases (44 associated with stavudine therapy), with a 25% mortality rate among the 36 patients with documented follow-up.45 Overall, the mortality associated with NRTI-associated lactic acidosis remains unacceptably high, at ∼50%.43
An “intermediate” hyperlactatemia syndrome has also been described, characterized by symptomatic hyperlactatemia or hepatic steatosis without systemic acidosis. This syndrome is almost uniformly associated with stavudine therapy, with an incidence of ∼13 per 1000 person-years.46,47 Of importance, there is evidence that lactate levels and symptoms can be controlled following modification of NRTI therapy to zidovudine or abacavir.48
With regard to the pathogenesis of hyperlactatemia syndromes, evidence has been provided from studies using exogenous lactate loading that increased lactate production is the fundamental defect in chronic asymptomatic hyperlactatemia, rather than decreased clearance of lactate (which primarily involves the liver).49 Indeed, lactate clearance appears to be upregulated as a compensatory response to an elevated systemic lactate load.49 The source of this excessive lactate production is unknown, although a carefully conducted exercise physiology study indicates that skeletal muscle is an unlikely candidate.50 By contrast, lactic acidosis represents a complete failure of lactate homeostasis, in which lactic acid accumulation progresses inexorably while NRTI therapy is maintained. In this scenario, the prominent role of liver mitochondrial dysfunction (indicated by the presence of microvesicular hepatic steatosis)37,43,46 leading to a shift from hepatic lactate consumption to lactate production would be predicted to have dramatic consequences for systemic lactate metabolism.
LIPOATROPHY
There is now strong evidence that the choice and duration of NRTI drugs represents the dominant risk factor for developing pathological loss of fat tissue, referred to as lipoatrophy. The importance of treatment choices in determining the risk of developing lipoatrophy is highlighted by the fact that this long-term complication is extremely rare in the general population as well as in treatment-naïve HIV-infected individuals.51 Hence, there are no known modifiable risk factors for developing lipoatrophy that are present in the general population. This may be contrasted with other aspects of the broader clinical syndrome commonly referred to as “lipodystrophy”, which also includes metabolic complications such as dyslipidemia and insulin resistance, and visceral/abdominal fat accumulation that often accompanies these metabolic changes52 (see Chapters 70 and 72). These metabolic complications of therapy, which have a much stronger association with HIV PI therapy than with NRTI drugs, are also highly prevalent in the general population, suggesting that risk may be modified by “nondrug” factors such as diet and physical activity. This topic will not be addressed here.
Clinical studies have demonstrated that NRTI therapy alone provides sufficient conditions for the development of lipoatrophy53–55 and that NRTI therapy is an independent risk factor for its occurrence in HAART-treated individuals.56–58 Clinical trials data have now confirmed the findings of observational cohort studies59 that stavudine therapy is associated with approximately twofold increased risk of lipoatrophy compared with zidovudine, so that the risk of clinically apparent lipoatrophy over 30 months is ∼10–20% in zidovudine-treated individuals, and 40–50% among those treated with stavudine.60–62 Significant differences in limb fat between stavudine and zidovudine treatment arms in the ACTG 384 trial have also been documented from 48 weeks onwards.63 Finally, switching NRTI therapy from stavudine to abacavir in an observational study64 and more recently in two randomized trials65,66 has been associated with significant improvements in fat wasting. In contrast, over 30 trials investigating HIV PI discontinuation as a therapeutic strategy67 have failed to demonstrate beneficial effects on fat wasting, although metabolic abnormalities such as dyslipidemia and insulin resistance did improve. It is notable, however, that improvement in fat wasting up to 96 weeks appear to be modest at best, with absolute increases in limb fat of ∼1%.65 Therefore, it would seem prudent to focus on identifying HAART regimens with a low risk of inducing fat wasting, since restoration of established fat wasting appears to be a slow, and possibly incomplete process.
The histopathological correlates of this syndrome also indicate that adipose tissue-specific mitochondrial toxicity is central to lipoatrophy pathogenesis. At the ultrastructural level, abnormal mitochondrial architecture and organellar proliferation are prominent features,68–70 while increased adipocyte apoptosis associated with lipoatrophy has been shown to improve after discontinuing stavudine therapy in favor of abacavir,71 but not after switching HIV PI therapy.72 Mitochondrial DNA depletion in adipose tissue samples has also been documented using semiquantitative techniques,70,73 and in studies of adipose tissue using precise real-time polymerase chain reaction quantitation methods which have demonstrated significant mitochondrial DNA depletion particularly associated with stavudine compared with zidovudine.74,75 Stavudine treatment has also been associated with more severe adipocyte mitochondrial DNA depletion and fat wasting over time compared with zidovudine therapy,76 as well as more severe effects on mitochondrial function.77
The role of NRTI drugs other than stavudine and zidovudine as risk factors for lipoatrophy has been more difficult to discern, particularly as lamivudine and didanosine are generally used in combination with these thymidine analog drugs. Thus far, no independent effect of these “second” NRTI drugs has emerged, with results from the available clinical trials completed to 30 months showing similar risk of lipoatrophy if stavudine is combined with didanosine or with lamivudine.60–62 Data regarding the concurrent use of zidovudine and didanosine is more difficult to obtain, although an early study of dual NRTI therapy by Saint-Marc and colleagues included a number of patients on didanosine with stavudine (d4T/ddI = 13/27, 48%) and zidovudine treatment groups (AZT/ddI = 13/16, 81%) that were well-matched for NRTI therapy duration.53 In this study, use of stavudine remained the most significant risk factor for lipoatrophy (relative risk 1.95 compared with zidovudine), while no significant didanosine effect could be demonstrated. Use of didanosine compared with lamivudine also had no significant effect on adipose tissue mitochondrial DNA depletion in a recent analysis of 232 biopsy samples, after adjustment for the effect of stavudine treatment.75 In terms of the newer drugs abacavir (an NRTI) and tenofovir (a nucleotide reverse transcriptase inhibitor), which would be predicted to have reduced predisposition to cause mitochondrial toxicity,30,31 clinical trials data show no evidence of an association with lipoatrophy. Indeed, 144-week data from the Gilead 903 study involving comparisons of tenofovir and stavudine therapy (combined with lamivudine and efavirenz in both arms), objectively measured limb fat was reduced by ∼40% in the stavudine arm compared with those on tenofovir.78
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


