Fig. 32.1
Clinical manifestations
Hyperglycemia develops as a result of three processes: increased gluconeogenesis, accelerated glycogenolysis, and impaired glucose utilization by peripheral tissues. Transient insulin resistance occurs due to hormone imbalance as well as elevated free fatty acids concentrations [4]. The combination of insulin deficiency and increased counter-regulatory hormones in DKA leads to the release of free fatty acids into the circulation from adipose tissue and to unrestrained hepatic fatty acid oxidation in the liver to ketone bodies, with resulting ketonemia and acidosis.
Clinical Manifestations
The clinical manifestations of DKA include the following:
Signs of dehydration: delayed capillary refill, postural changes of blood pressure and pulse, dry mucous membranes.
Signs of acidosis: deep-sighing respirations (Kussmaul) in attempt to blow off carbon dioxide, shortness of breath, chest pain due to accessory muscle exhaustion.
Results of osmotic diuresis and vomiting, dehydration, and hyperosmolality: abdominal pain mimicking pancreatitis or an acute surgical abdomen.
Results of counter-regulatory hormone release: elevated leukocyte count to 15,000–20,000/mm3.
Signs of hyperosmolality: progressive obtundation and loss of consciousness related to the degree of evolving hyperosmolality.
In DKA, metabolic decompensation usually develops over a period of hours to a few days. Patients with DKA classically present with lethargy and a characteristic hyperventilation pattern with deep slow breaths (Kussmaul respirations) associated with the fruity odor of acetone. They complain of nausea and vomiting, and abdominal pain is somewhat less frequent. The abdominal pain can be quite severe and may be associated with distention, ileus, and tenderness without rebound but usually resolves relatively quickly with therapy unless there is underlying abdominal pathology. Most patients are normotensive, tachycardic, and tachypneic and have signs of mild to moderate dehydration. Hypothermia has been described in DKA, and patients with underlying infection might not manifest fever. Patients with DKA can have stupor and obvious profound dehydration, and they often demonstrate focal neurologic deficits such as Babinski reflexes, asymmetrical reflexes, cranial nerve findings, paresis, fasciculations, and aphasia [1].
Laboratory Test Results and Differential Diagnosis
Laboratory tests that are routinely monitored in DKA include hemoglobin, white blood cell and differential count, glucose, electrolytes, arterial blood gases, ketones, blood urea nitrogen (BUN), and creatinine. The severity of DKA is classified as mild, moderate, or severe based on the severity of metabolic acidosis (blood pH, bicarbonate, and ketones) and the presence of altered mental status [4].
Acidosis is always present in DKA, and the serum HCO3 concentration is usually less than 10 mEq/L. The acidosis is due to production and accumulation of ketones in the serum. Three ketones are produced in DKA: two ketoacids (β-hydroxybutyrate and acetoacetate) and the neutral ketone acetone. Ketones can be detected in serum and urine using the nitroprusside reaction on diagnostic strips for use at the patient’s bedside or in the clinical laboratory. This test detects acetoacetate more effectively than acetone and does not detect an increased concentration of β-hydroxybutyrate. Particularly in severe DKA, β-hydroxybutyrate is the predominant ketone, and it is possible although unusual to have a negative serum nitroprusside reaction in the presence of severe ketosis. However, under these circumstances the serum HCO3 is still markedly reduced and the anion gap is increased, indicating metabolic acidosis. The urinary β-hydroxybutyrate can be measured at many centers and commercially but is not usually readily available.
The anion gap is a readily available index for unmeasured anions in the blood (normal <14 mEq/L): Anion gap = sodium − (chloride + bicarbonate).
Most patients with DKA present with an anion gap greater than 20 mEq/L, and some present with a gap greater than 40 mEq/L. However, occasional patients have a hyperchloremic metabolic acidosis without a significant anion gap [5].
Patients with DKA almost invariably have large amounts of ketones in their urine. The serum glucose in DKA is usually in the 500 mg/dL range. However, an entity known as euglycemic DKA has been described, particularly in the presence of decreased oral intake or in pregnancy, in which the serum glucose is normal or near normal but the patient requires insulin therapy for the clearance of ketoacidosis [6]. The arterial pH is commonly less than 7.3 and can be as low as 6.5. There is partial respiratory compensation with hypocarbia. Patients are often mildly hyperosmolar, although osmolalities greater than 330 mOsm/kg are unusual without mental status changes (Table 32.1).
Table 32.1
Typical laboratory findings in DKA
Average | Range | |
|---|---|---|
Plasma glucose | 600 mg/dL | 200–2,000 mg/dL |
Plasma B-hydroxybutyrate | 14 mmol/L | 4–20 mmol/L |
Plasma HCO3 | 10 mEq/L | 4–18 mEq/L |
Blood pH | 7.15 | 6.80–7.30 |
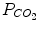 | 20 mmHg | 14–30 mmHg |
Plasma anion gap (Na-[Cl + HCO3]) | 29 | 16–35 mEq/L |
Differential Diagnosis
The following causes of metabolic acidosis need to be considered in the differential diagnosis of DKA.
Lactic acidosis is the most common cause of metabolic acidosis in hospitalized patients and can be seen in patients with uncomplicated diabetes as well as those with DKA. Lactic acidosis usually occurs in the setting of decreased tissue oxygen delivery, resulting in the nonoxidative metabolism of glucose to lactic acid. Lactic acidosis complicates other primary metabolic acidoses as a consequence of dehydration or shock, and assessing its relative contribution can be difficult. The presentation is similar to that of DKA. In pure lactic acidosis, the serum glucose and ketones should be normal and the serum lactate concentration should be greater than 5 mm. The therapy of lactic acidosis is directed at the underlying cause and optimizing tissue perfusion [7].
Starvation ketosis is caused by inadequate carbohydrate availability, resulting in physiologically lipolysis and ketone production to provide fuel substrates for muscle. The blood glucose is usually normal. The urine can have large amounts of ketones, but the blood rarely does. Arterial pH is normal, and the anion gap is at most mildly elevated.
Alcoholic ketoacidosis is a more severe form of starvation ketosis wherein the appropriate ketogenic response to poor carbohydrate intake is increased through as yet poorly defined effects of alcohol on the liver. Classically, these patients are long-standing alcoholics for whom ethanol has been the main caloric source for days to weeks. The ketoacidosis occurs when for some reason alcohol and caloric intake decreases. In isolated alcoholic ketoacidosis, the metabolic acidosis is usually mild to moderate. The anion gap is elevated. Serum and urine ketones are always present. However, alcoholic ketoacidosis produces an even higher ratio of β-hydroxybutyrate to acetoacetate than DKA does, and negative or weakly positive nitroprusside reactions are common. Respiratory alkalosis associated with delirium tremens, agitation, or pulmonary processes often normalizes the pH but should be evident with careful analysis of acid–base status. Usually, the patient is normoglycemic or hypoglycemic, although mild hyperglycemia is occasionally present. Patients who are significantly hyperglycemic should be treated as if they have DKA. The therapy of alcoholic ketoacidosis consists of thiamine, carbohydrates, fluids, and electrolytes, with special attention to the more severe consequences of alcohol toxicity, alcohol withdrawal, and chronic malnutrition [8].
Uremic acidosis is characterized by extremely large elevations in the BUN (often >200 mg/dL) and creatinine (>10 mg/dL) with normoglycemia. The pH and anion gap are usually only mildly abnormal. The treatment is supportive, with careful attention to fluid and electrolytes until dialysis can be performed. Rhabdomyolysis is a cause of renal failure in which the anion gap can be significantly elevated and acidosis can be severe. There should be marked elevation of creatine phosphokinase and myoglobin. Mild rhabdomyolysis is not uncommon in DKA, but the presence of hyperglycemia and ketonemia leaves no doubt about the primary etiology of the acidosis [9].
Toxic ingestions can be differentiated from DKA by history and laboratory investigation. Salicylate intoxication produces an anion gap metabolic acidosis usually with a respiratory alkalosis. The plasma glucose is normal or low, the osmolality is normal, ketones are negative, and salicylates can be detected in the urine or blood. Salicylates can cause a false-positive glucose determination when using the cupric sulfate method and a false-negative result when using the glucose oxidase reaction detection of ketoacids [10].
When DKA is considered, the diagnosis can be made quickly with routine laboratory tests. Blood and urine glucose and ketones can be obtained in minutes with glucose oxidase-impregnated strips and the nitroprusside reaction, respectively.
Osmolality
The increase in osmolality (mOsm/L) that occurs in DKA must be differentiated from the increase in osmolality seen in hyperosmolar-hyperglycemic nonketotic (diabetic) coma (HHNC). The osmolality can be estimated using the following formula: Osmolality = (2 × sodium) + (glucose/18).
Patients with DKA rarely present with hyperosmolality and coma. In HHNC, the osmolarity is generally greater than 350 mOsm/L and can exceed 400 mOsm/L. The serum sodium and potassium can be high, normal, or low and do not reflect total-body levels, which are uniformly depleted. The glucose is usually greater than 600 mg/dL, and levels higher than 1,000 mg/dL are quite common. In pure HHNC, there is not a significant metabolic acidosis or anion gap.
Patients often present with combinations of the preceding findings. HHNC can involve mild to moderate ketonemia and acidosis. Alcoholic ketoacidosis can contribute to either DKA or HHNC. Lactic acidosis is common in severe DKA and HHNC.
Present Therapy [11]
The general approach is to provide necessary fluids to restore the circulation, treat insulin deficiency with continuous insulin, treat electrolyte disturbances, observe the patient closely and carefully, and search for underlying causes of metabolic disturbances.
Fluids
The primary goal in the initial management of DKA is to restore intravascular volume and improve tissue perfusion, and the secondary goal is to maintain a brisk diuresis. This will decrease insulin counter-regulatory hormones levels and glucose concentration [1]. Fluid replacement alone may decrease serum glucose concentration by as much as 23 % through increased renal perfusion and loss of glucose in the urine [1, 12]. Initial intravenous fluids should be given rapidly to achieve hemodynamic stability, then decreased to rate that allows for replacement of the total deficit over a 24-h period. The total body water deficit (TBW) in these patients is usually 5–10 L. The total body water deficit may be calculated by the formula: TBW deficit (L) = 0.6 × wt (kg) + [1 − 140/serum sodium]. The goal in fluid administration is to replace approximately 50 % of TBW deficit in the first 8 h and the remainder in the subsequent 16 h. The initial fluid of choice is normal saline (0.9 % sodium chloride) with 1 or 2 L administered in the first hour. The subsequent choice for fluid replacement depends on the patient’s hydration status, serum electrolytes levels, and urinary output [11]. One approach suggest an infusion rate of 0.9 % NaCl 500 mL/h until hemodynamically stable. Then the rate is decreased to 250 mL/h and can be switched to half normal saline to replace the large free water deficit [1].
When the blood glucose level falls below 250 mL/dL, a solution of 5 % dextrose should be added to the intravenous fluid. This allows for continued administration of insulin to treat the ketosis and acidosis without causing hypoglycemia. The serum glucose should be maintained between 150 and 200 mg/dL until the ketoacidosis has resolved. Dextrose administration may be increased to 10 or 20 % if glucose levels remain below 100 mg/dL.
Special care should be taken to avoid overhydration in children, patients with cardiac or renal compromise, and elders with DKA. The lung sounds and oxygenation should be assessed frequently. In children, mental status may be the first sign of cerebral edema [11].
Insulin
Insulin therapy will improve the hyperglycemia, ketosis and acidosis that occur in DKA. In the past, high doses of insulin (upward of 50 U/h) were favored. In later studies, low-dose insulin therapy (0.1 U/kg per hour) has been shown to be as effective as higher doses in producing a decrease in serum glucose and clearance of ketones. Furthermore, low-dose therapy results in a reduction in the major morbidity of intensive insulin therapy: hypoglycemia and hypokalemia.
Studies have also shown that intravenous insulin is significantly more effective than intramuscular or subcutaneous insulin in lowering the ketone body concentration over the first 2 h of therapy. The subcutaneous route is probably inappropriate for the critically ill patient because of the possibility of tissue hypoperfusion and slower kinetics of absorption; however, a study has documented that subcutaneous rapid-acting insulin analogue administered every 1–2 h was as safe and effective as intravenous regular insulin in the treatment of patients with uncomplicated DKA [13]. Numerous studies attest to the efficacy of intramuscular therapy in severe DKA. When there is insufficient nursing monitoring or intravenous access to allow safe intravenous administration, intramuscular therapy would be the route of choice.
Lastly, it has been shown that a 10-U intravenous insulin priming dose when insulin therapy is started significantly improves the glycemic response to the first hour of therapy. The rationale is to saturate insulin receptors fully before beginning continuous therapy and to avoid the lag time necessary to achieve steady-state insulin levels. Because insulin adsorbs to intravenous tubing, 50 mL of the infusion should be run through the pump before beginning the infusion [11].
In the rare instances in which the glucose does not decrease at least 10 % or 50 mg/dL in an hour, the insulin infusion rate should be increased by 50–100 % and a second bolus of intravenous insulin should be administered. As the glucose level decreases, it is usually necessary to decrease the rate of infusion. After the glucose reaches approximately 250 mg/dL, it is prudent to decrease the insulin infusion rate and administer dextrose. It usually takes an additional 12–24 h to clear ketones from the circulation after hyperglycemia is controlled. With resolution of ketosis, the rate of infusion approaches the physiologic range of 0.3–0.5 U/kg per day. Criteria for the resolution of DKA includes glucose less than 200 mg/dL, serum bicarbonate at least 18 mmol/L, and a venous pH greater than 7.3 [11].
When the decision is made to feed the patient, the patient should be switched from intravenous or intramuscular therapy to subcutaneous therapy. Subcutaneous insulin should be administered before a meal and the insulin drip discontinued approximately 2 h later. The glucose should be checked in 2 h and at least every 4 h subsequently until a relatively stable insulin regimen is determined. Early conversion to oral feeding and subcutaneous insulin therapy is associated with a shorter hospital stay.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


