Pain
Motor regression
Weakness
Gait abnormality/deterioration
Torticollis
Progressive kyphoscoliosis
Hydrocephalus
Sphincter disturbance
Reflex changes
Sensory impairment
10.2.3.1 Diagnostic Imaging
MRI is the diagnostic tool of choice for all spinal tumors (Miyazawa et al. 2000; Sun et al. 2003). There is virtually no role for plain radiographs or computed tomography (CT) images because of the associated radiation exposure and limited anatomic detail seen. Astrocytomas are commonly located eccentrically within the spinal cord, and there is often heterogeneous contrast enhancement following injection of gadolinium (Baleriaux 1999; Osborn 1994) (Figs. 10.3 and 10.4). Diffusion tensor imaging (DTI) can be helpful in defining the relationship of the lesion to critical spinal pathway. In a series of ten patients, tractography was capable of demonstrating fiber splaying/displacement versus pathway infiltration. This information contributed to the decision between aggressive resection compared to debulking and biopsy (Choudhri et al. 2014). DTI and perfusion-weighted imaging (PWI) can be helpful in differentiating IMSCTs from other tumorlike lesions in the cervical cord (Liu et al. 2014). Approximately 75 % of astrocytomas occur in the cervicothoracic region, 20 % in the distal spinal cord, and 5 % in the filum terminale (Osborn 1994). Unlike ependymomas, which typically span three to four vertebral bodies, spinal cord astrocytomas are more extensive spanning several levels to holocord (Baleriaux 1999; Osborn 1994; Ebner et al. 2012). Although MRI has improved our ability to identify the exact location of IMSCTs, a precise histopathologic diagnosis requires tissue biopsy (Kopelson and Linggood 1982; McCormick et al. 1990a; Hulshof et al. 1993; Minehan et al. 1995; Lee et al. 1996; Innocenzi et al. 1997; Jallo et al. 2001).
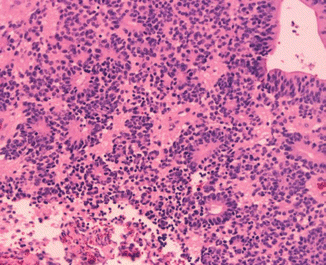
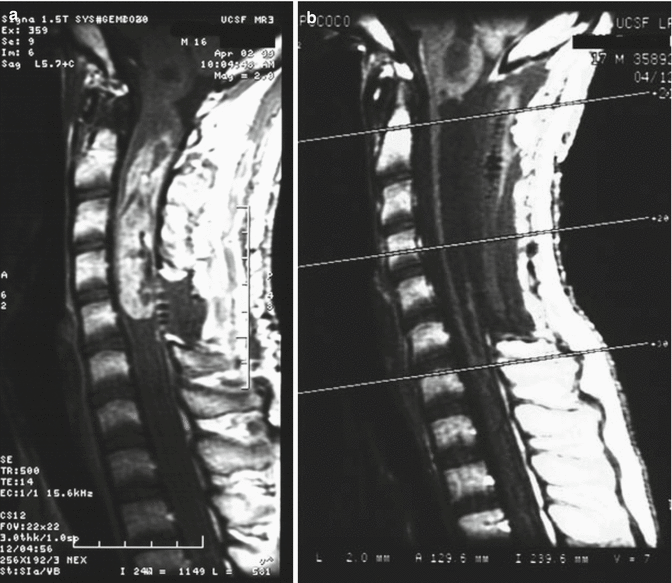

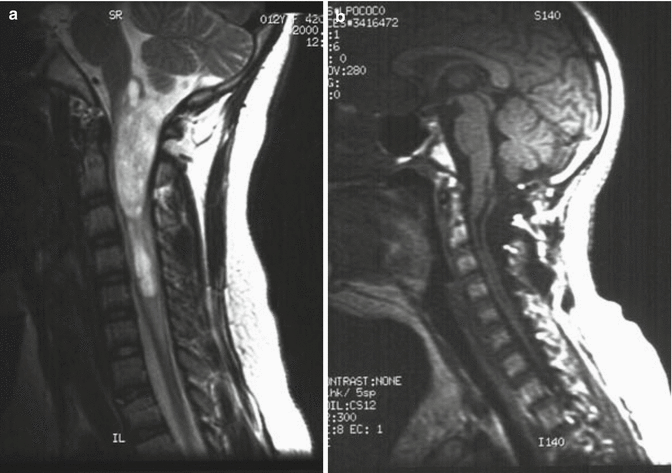
Fig. 10.1
Histological features of ependymoma. This image illustrates the ependymal rosettes which are formed from columnar cells arranged around a central lumen. Also in the top right hand corner, a pseudorosette, cells arranged radially around a blood vessels, can be appreciated
Fig. 10.2
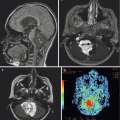
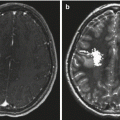
A 17-year-old male presented with left-arm numbness and tingling. (a) The preoperative MRI scan reveals an intramedullary cervical cord mass in the sagittal T1-weighted image with contrast. Gross total resection was achieved, and pathology was consistent with a grade II ependymoma. (b) A postoperative MRI showed resection of the mass with no evidence of residual tumor as demonstrated in the sagittal T1-weighted image with contrast
Fig. 10.3
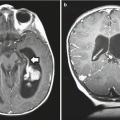
A 3-year-old girl presented with 6 months of intermittent, worsening back pain. (a) The MRI scan revealed a large intramedullary mass extending from T3 to T7, shown here in a sagittal T2-weighted image. The histology was consistent with pilocytic astrocytoma. (b) The postoperative MRI scan demonstrates removal of the centrally located tumor. Nodular enhancement in the area of the surgery is seen in a sagittal T2-weighted image. Although nodular enhancement was present in the first imaging, subsequent MRIs showed complete resolution 6 months after resection
Fig. 10.4
A 12-year-old girl presented with a 3-month history of progressive right arm weakness and clumsiness. (a) The preoperative MRI scan revealed an intramedullary spinal cord tumor extending from the cervicomedullary junction to C4, as shown in sagittal T2-weighted image. Histology was consistent with a pilocytic astrocytoma. (b) The postoperative MRI demonstrates that the enhancing mass has been resected, as shown in the sagittal T1-weighted image
10.3 Ependymoma
10.3.1 Epidemiology
Ependymomas are thought to arise from the ependymal lining of the ventricles and central canal and can occur both in the brain and spinal cord. The majority of ependymomas are sporadic, but they can also be associated with NF2. In children, ependymomas usually arise in the cervical region and occur less frequently than astrocytomas (McCormick et al. 1990b; Brotchi et al. 1991; Fine et al. 1995; Goh et al. 1997; Miller 2000; Schwartz and McCormick 2000; Hanbali et al. 2002). Miller identified only 16 ependymomas out of 117 (14 %) cases of pediatric IMSCTs (Miller 2000). Although intramedullary ependymomas are the most common spinal cord tumor in adults (Mork and Loken 1977; Sonneland et al. 1985; Helseth and Mork 1989; Whitaker et al. 1991; Clover et al. 1993; Hulshof et al. 1993; Hoshimaru et al. 1999; Schwartz and McCormick 2000; Chang et al. 2002; Hanbali et al. 2002; Parsa et al. 2004), they are less common in the pediatric population (Constantini et al. 1996; Miller 2000; Constantini 1996) In their series of pediatric IMSCTs, Constantini and colleagues did not find any ependymomas in children less than 3 years of age (Constantini et al. 1996).
10.3.2 Pathology
10.3.2.1 Grading
The World Health Organization (WHO) classification of CNS tumors (Kleihues et al. 2002; Louis et al. 2007) divides ependymomas into four types: subependymoma (grade I), myxopapillary ependymoma (grade I), benign or “classic” ependymoma (grade II), and anaplastic ependymoma (grade III). Subependymomas are considered benign, slow-growing, and intraventricular tumors and have a good prognosis, although they are rarely encountered in the spinal cord. Myxopapillary ependymomas are unique tumors because they usually arise from the filum terminale or conus medullaris (Sonneland et al. 1985). Nearly all are histologically benign and are associated with a good long-term survival (Mork and Loken 1977; Cooper 1989; McCormick et al. 1990b; Epstein et al. 1993; Chang et al. 2002; Hanbali et al. 2002; Russell 1989). Although most spinal cord ependymomas in children are grade II tumors, anaplastic ependymomas do occur infrequently and are believed to arise from the malignant transformation of lower-grade tumors (Kleihues et al. 2002).
10.3.2.2 Histopathology
Subependymomas are characterized by clusters of glial cells in a dense fibrillary matrix and are often associated with small cysts. Ependymomas are highly cellular tumors, irrespective of their grade. Myxopapillary ependymomas are characterized by cuboidal or elongated tumor cells arranged in a papillary and radial pattern around the vascular and stromal cores. Little mitotic activity is present, but a matrix of abundant mucin can accumulate between myxopapillary ependymoma cells and vessels.
The gross appearance of grade II spinal cord ependymomas is that of a soft, red or grayish-purple, somewhat friable mass (McCormick et al. 1990b; Schwartz and McCormick 2000; Sun et al. 2003). Cystic degeneration and hemorrhage are common in these vascular tumors (Sun et al. 2003). Although unencapsulated, these tumors are usually well circumscribed and do not infiltrate adjacent spinal cord tissue (Goh et al. 1997; Parsa et al. 2004; Parsa and McCormick 2005). Microscopic features include pseudorosettes and perivascular clustering and cuffing and immunoreactivity for glial fibrillary acidic protein (GFAP). Pseudorosettes are formed by clustering of cuboidal or columnar cells in a radial pattern around the blood vessels (Fig. 10.1). True rosettes, which appear as a ring of several nuclei from which interlacing neurofibrils converge in the center, can also be present (Schwartz and McCormick 2000). Mitotic figures are rare, but an occasional nonpalisading focus of necrosis can be found in low-grade ependymomas. As measured by MIB-1 immunohistochemistry, the proliferative activity of spinal cord ependymoma is significantly lower than that of intracranial ependymoma. Proliferative indices greater than 2.0 % may be associated with an increased risk of recurrence (Iwasaki et al. 2000). The atypical variants clear cell ependymoma and tanycytic ependymoma can mimic oligodendroglioma and astrocytoma, respectively (Goh et al. 1997).
Anaplastic ependymomas differ from grade II ependymomas. While grade II ependymomas morphologically appear similar to nonneoplastic ependymal cells, anaplastic ependymomas demonstrate clear evidence of malignancy such as increased mitotic activity, increased cellularity with microvascular proliferation, and pseudopalisading necrosis. Anaplastic ependymomas can be extremely invasive and are poorly differentiated.
10.3.2.3 Molecular Biology and Genetics
Myxopapillary ependymomas have a much higher propensity for aneuploidy or polyploidy, especially of chromosome 7, when compared to other ependymomas (Gilhuis et al. 2004; Santi et al. 2005). Anaplastic ependymomas (WHO grade III) of the spinal cord are rare, and genetic alterations remain largely undefined (Ebert et al. 1999).
Molecular and genetic events associated with spinal ependymoma have been described. Ebert and colleagues analyzed 62 ependymal tumors, including myxopapillary ependymomas, subependymomas, classic ependymomas, and anaplastic ependymoma. They showed allelic loss of chromosomes 10q (5 out of 56) and 22q (12 out of 54) (Ebert et al. 1999). Somatic mutations of the NF2 gene were detected in six of the tumors examined, and in each case the tumor was from a grade II spinal cord ependymoma. These results were confirmed by another group which also found mutations in the NF2. In addition, loss of heterozygosity (LOH) of 22q was present in all spinal intramedullary ependymomas (n = 6) (Lamszus et al. 2001). Allelic loss on 22q was also frequently observed and was more common in intramedullary spinal ependymomas than in tumors in other locations (Lamszus et al. 2001).
In a report of 22 pediatric ependymomas, LOH at chromosome 22 was observed in two cases, deletions of chromosome 17 in another two cases, and the deletion or rearrangement of chromosome 6 in another five cases (Kramer et al. 1998). In addition, a low-penetrance ependymoma susceptibility locus has been mapped to chromosome 22q11 (Hulsebos et al. 1999; Ammerlaan et al. 2005), suggesting the role of alternative predisposing genes apart from NF2.
Overall, 75 % of all ependymomas display chromosomal aberrations or rearrangements over several different chromosomes, the most frequent LOHs being found on the long arms of chromosomes 6 (30.3 %), 9 (27.3 %), and 17 (Huang et al. 2003). In 18 pediatric ependymomas, von Haken and colleagues reported a 50 % incidence of allelic mutations on the short arm of chromosome 17 (von Haken et al. 1996). LOH was also detected on 3p14 (13.3 %), 10q23 (10.3 %), and 11q (18.2 %). Monosomy of chromosome 22 is present in approximately 30 % of ependymomas (Scheil et al. 2001), with aberrations or alterations of 22q existing in up to 40 % of all ependymomas.
Another distinction between spinal and cranial ependymoma may lie in the methylation of particular tumor-related genes. A study examining the methylation of a putative tumor suppressor gene, HIC–1 on chromosome 17p13.3, showed a significant correlation between hypermethylation of HIC–1 and cranial localization (p = 0.019, n = 52) (Waha et al. 2004). Losses in chromosomes 1p and 16q, which occur in other CNS tumors, have not been found in ependymoma (Bijlsma et al. 1995). The apparent genetic differences between ependymomas in the brain and those in the spine suggest that different molecular mechanisms exist that lead to the pathogenesis of each. Because primary brain and spine tumors are rarely, if ever, associated with each other, these distinctions may indicate the need to reclassify spinal ependymoma separately from intracranial ependymoma.
10.3.2.4 Association with Neurofibromatosis Type 2
NF2 is a rare autosomal dominant genetic disorder associated with tumors of the CNS (see Chap. 12) (Mulvihill et al. 1990). Its prevalence is 1 in 40,000 individuals (Evans et al. 1992), and is caused by a mutation of the NF2 tumor suppressor gene (also known as merlin or schwannomin) located on chromosome 22 (Rouleau et al. 1987, 1993; Trofatter et al. 1993). Patients with NF2 have a high incidence of several CNS tumors, including vestibular schwannomas and meningiomas (Martuza and Eldridge 1988). Several authors have also noted an association between NF2 and intramedullary spinal cord ependymomas (Martuza and Eldridge 1988; Rodriguez and Berthrong 1966; Mautner et al. 1993; Lee et al. 1996; Lamszus et al. 2001; Egelhoff et al. 1992). NF2 patients represent approximately 2.5 % of patients with IMSCTs, yet only 0.03 % of the population (Lee et al. 1996). In addition, in one small study, 71 % of patients with intramedullary spinal cord ependymomas and no other clinical features of NF2 were shown to possess mutations in the NF2 gene (Birch et al. 1996). More recently, Garcia and Guttman investigated the mechanism by which the NF2 protein Merlin regulates spinal neural differentiation and glial proliferation. They demonstrated that Merlin negative regulates these cell functions in a manner dependent on ErbB2, and they further observed increased Erb2 activation in NF2-associated ependymomas; they further hypothesize that ErbB2 may be a rational therapeutic target for medical therapy for NF2-associated spinal ependymoma (Garcia et al. 2014).
10.3.3 Clinical Features
Arising from ependymal cells lining the central canal, intramedullary ependymomas are well circumscribed, slow-growing tumors usually located in the center of the cervical spinal cord and cause symmetric expansion of the cord (McCormick et al. 1990a; Brotchi et al. 1991; Fine et al. 1995; Goh et al. 1997; Miller 2000; Schwartz and McCormick 2000; Hanbali et al. 2002). Patients typically complain of dysesthesia correlating to the level of the tumor for months to years prior to diagnosis. Other symptoms include paresthesia, radicular pain, bowel and bladder dysfunction, and other sensory disturbances (Rawlings et al. 1988; McCormick and Stein 1990; McCormick et al. 1990b; Clover et al. 1993; Epstein et al. 1993; Hulshof et al. 1993; Asazuma et al. 1999; Hoshimaru et al. 1999; Schwartz and McCormick 2000; Chang et al. 2002; Hanbali et al. 2002; Peker et al. 2004; Shrivastava et al. 2005). Children most often present with pain, weakness, gait abnormality, torticollis, or progressive kyphoscoliosis (Constantini et al. 1996, 2000). Hydrocephalus also is more common in pediatric patients with intramedullary spinal cord ependymomas than in adult patients and may require cerebrospinal fluid (CSF) shunting (Houten and Weiner 2000; Houten and Cooper 2000). A sudden decline in neurologic function may occur following intratumoral hemorrhage (McCormick et al. 1990b). Motor impairment usually occurs late in the disease progression as the expanding tumor thins the surrounding spinal cord to a few millimeters (Epstein et al. 1993) (Table 10.1). This differs from intramedullary astrocytomas, which tend to present with pain and progressive motor dysfunction over a shorter time (Epstein et al. 1993).
10.3.4 Diagnostic Imaging
The anatomic features of spinal cord tumors are best evaluated with magnetic resonance imaging (MRI) (Miyazawa et al. 2000; Sun et al. 2003). Intramedullary spinal cord ependymomas are typically centrally located lesions with sharply defined rostral and caudal margins, enhancing borders, and typically spanning three to four vertebral body segments (Baleriaux 1999; Miyazawa et al. 2000). Spinal cord ependymomas commonly demonstrate symmetric enlargement of the spinal cord, unlike astrocytomas, which exhibit a nodular or asymmetric pattern of growth (Kopelson and Linggood 1982; McCormick et al. 1990a; Hulshof et al. 1993; Minehan et al. 1995; Lee et al. 1996; Innocenzi et al. 1997; Iwasaki et al. 2000; Miyazawa et al. 2000; Jallo et al. 2001).
Spinal cord ependymomas are isointense on T1-weighted MR images and slightly hyperintense on T2-weighted MR images (Miyazawa et al. 2000; Sun et al. 2003) (Table 10.2). However, signal heterogeneity can occur with cyst formation, necrosis, or hemorrhage (Miyazawa et al. 2000). A “cap sign” is typically associated with spinal cord ependymomas and represents areas of low signal density on either border of the tumor mass itself. This “cap” hypointensity at the tumor margin is often due to hemosiderin deposits from secondary, chronic hemorrhage (Baleriaux 1999; Miyazawa et al. 2000; Chang et al. 2002). Almost all intramedullary ependymomas enhance with contrast, but to a lesser degree than intracranial ependymomas (Sun et al. 2003) (Fig. 10.2). Occasionally, these spinal cord ependymomas can present with subarachnoid hemorrhage.
Table 10.2
Magnetic resonance imaging of intramedullary spinal cord tumors
Ependymomas | Astrocytomas | |
|---|---|---|
Location | Centrally located; mostly in the cervical spine but in children also present in the conus | Eccentrically located, usually widens the spinal cord, 75 % of astrocytomas in the cervical and thoracic regions, 20 % in the distal cord, 5 % in the filum terminale |
T1 | Isointense/hypointense | Isointense/hypointense |
T1 with contrast | Axial view – cord symmetrically expanded. Enhances with contrast but less than astrocytomas | Ill-defined borders axial view – cord asymmetrical, “lumpy”; heterogeneous, moderate, partial contrast enhancement |
Spinal cord ependymoma-related cysts are common and are classified into three types: cystic tumors from tumor necrosis and hemorrhage, syrinx formation from disturbances of CSF formation, and rostral and caudal cysts from reactive products of IMSCTs (Sun et al. 2003). Ependymoma-associated cysts appear hypointense on T1-weighted MR images and hyperintense on T2-weighted images (Sun et al. 2003). These cysts are also centrally located and cause symmetric expansion of the spinal cord (Sun et al. 2003). A tumor-associated syrinx has similar MR characteristics to CSF and is present in over 50 % of spinal cord ependymomas (Chang et al. 2002). Multivariate analysis has determined that the presence of syringohydromyelia strongly favors a diagnosis of ependymoma over astrocytoma (Kim et al. 2014). The majority of rostral and caudal cysts are also hypointense on T1-weighted MR images and hyperintense on T2-weighted MR images (Sun et al. 2003).
10.4 Von Hippel–Lindau Disease and Spinal Hemangioblastoma
Hemangioblastomas are benign (WHO grade I) vascular tumors predominantly found in the cerebellum and spinal cord (see Chap. 12). First described by Arvid Lindau as cystic lesions in the cerebellum, CNS hemangioblastomas are usually sporadic, but 20–30 % of cases occur in association with von Hippel–Lindau (VHL) disease (Glasker 2005). VHL is an autosomal dominant disorder with 90 % penetrance attributable to loss of a tumor suppressor gene on chromosome 3p25–26 (Kley et al. 1995). The VHL gene encodes for a protein required for oxygen-dependent degradation of hypoxia-inducible factor-1 alpha (HIF-1a). Dysfunction or absence of the VHL gene product leads to constitutive overexpression of HIF-1a, which then leads to increased levels of vascular endothelial growth factor (VEGF) and other proangiogenic signals (Kim and Kaelin 2004). Additional information is provided in Chap. 12.6.
Lesions associated with VHL include CNS hemangioblastoma, retinal angioma, renal cysts, renal cell carcinoma, pancreatic cysts, pheochromocytoma, and epididymal cystadenoma (Glavac et al. 1996). VHL families can be grouped according to the presence or absence of pheochromocytomas (Neumann et al. 1995). Nearly all families with pheochromocytomas have missense mutations of the VHL gene. Using tissue microdissection, Vortmeyer and colleagues have demonstrated consistent LOH at the VHL gene locus in the stromal cells, implicating these cells in the pathogenesis of hemangioblastoma (Vortmeyer et al. 1997).
CNS hemangioblastoma occurs in both type I (without pheochromocytoma) and type II (with pheochromocytoma) VHL disease. Common sites include the posterior fossa (80 %) and the spinal cord (20 %). VHL-related hemangioblastomas have been reported to harbor germline mutations (94 %) and LOH (62 %) at the VHL gene (Glasker et al. 1999, 2001; Glasker 2005). Over 150 different germline mutations have been identified and include deletion and missense and nonsense frameshift mutations. The resultant biallelic inactivation of the VHL gene suggests a “2-hit” model of tumorigenesis in VHL patients. VHL patients are usually heterozygous for the germline VHL mutant, and a “second hit” at the remaining wild-type VHL gene then causes neoplastic progression. In contrast, sporadic hemangioblastomas contain only 50 % LOH and 23 % germline mutations at the VHL gene, suggesting alternate pathways to biallelic inactivation and tumorigenesis in sporadic cases (Glasker 2005).
Other mutations and sites of LOH have been implicated in the development of sporadic hemangioblastomas. LOH of chromosome 22q13 was found in 5 of 8 patients with non-VHL-related hemangioblastoma, with only 3 of 8 patients harboring LOH at chromosome 3p21–23 (Beckner et al. 2004). Differences in the molecular and genetic origins of hemangioblastoma may indicate differences between patients with VHL disease and CNS hemangioblastomas and those with sporadic CNS hemangioblastomas.
10.5 Other Intramedullary Spinal Cord Tumors and Lesions
Inclusion tumors and cysts, metastases, nerve sheath tumors, neurocytoma, and melanocytoma account for much of the remainder of intramedullary mass lesions. Approximately 4 % of apparent IMSCTs are nonneoplastic lesions (Lee et al. 1998). Lipomas are the most common developmental lesion and account for about 1 % of all intramedullary spinal cord masses (Lee et al. 1998).
10.6 Treatment
Surgery is the treatment of choice for IMSCTs, and excellent results are associated with gross total resection (Houten and Weiner 2000; Iwasaki et al. 2000). Although the outcome for low-grade spinal cord astrocytomas is better in children than in adults, the prognosis for spinal cord astrocytomas is not as favorable as that of ependymoma (Goh et al. 1997; Houten and Weiner 2000; Iwasaki et al. 2000; Hanbali et al. 2002; Houten and Cooper 2000). Radical resection has been shown to prolong survival for non-disseminated WHO grades II and III astrocytomas, but must be weighed against the risk of causing neurologic deficits. GTR currently has no role in treatment of WHO grade IV tumors. Adjuvant radiotherapy is commonly used in cases of malignant astrocytomas or subtotally resected tumors. Little is known about the utility of chemotherapy for spinal cord astrocytoma or ependymoma.
10.6.1 Surgery
10.6.1.1 Surgical Principles
Surgery is effective for diffusely infiltrating spinal cord astrocytomas, and often a tissue diagnosis is all that can be safely accomplished (Houten and Weiner 2000; Houten and Cooper 2000). Pilocytic spinal cord astrocytomas, however, can be completely resected. The goal of surgery for intramedullary ependymoma is gross total resection (GTR) and preservation of neurologic function (Cooper 1989; McCormick et al. 1990a, b; McCormick and Stein 1990; Epstein et al. 1993; Cristante and Herrmann 1994; Chang et al. 2002; Peker et al. 2004). Ependymomas are typically non-infiltrative lesions that cause compression of the adjacent cord parenchyma, and the presence of a well-defined interface between the spinal cord and the tumor facilitates surgical resection (Sandalcioglu et al. 2005). An adequate myelotomy is necessary to fully expose the tumor and allow an accurate tissue diagnosis (Hanbali et al. 2002). An intraoperative frozen section diagnosis consistent with ependymoma should prompt an attempt at GTR. Conversely, identification of a malignant tumor requires that the surgeon carefully weigh the risks versus benefits of further resection, factoring in the WHO grade, intraoperative appearance, quality of tumor margins, and stability of neuromonitoring signals. The presence of a syrinx may improve the chances of a GTR, but it cannot be used as an independent predictor of outcome (Samii and Klekamp 1994; Chang et al. 2002; Peker et al. 2004).
10.6.1.2 Surgical Approach
Pediatric IMSCTs are approached by performing a laminectomy or, more commonly, an osteoplastic laminotomy. The osteoplastic technique has been associated with decreased rates of progressive kyphotic deformity requiring fusion (McGirt et al. 2008b). It involves removal of the bony lamina to expose the dura and spinal cord at the relevant levels as indicated by the preoperative MRI, followed by replacement of the posterior bony elements after the tumor resection is completed. To expose the dura, parallel cuts are made in the lamina of the involved spinal segments with either a high-speed side-cutting drill or rongeurs. The supraspinous and intraspinous ligaments are then sharply dissected at the caudal end of the laminoplasty flap prior to its elevation. Following tumor resection, the flap is resecured with sutures or plates (metal or absorbable). Preservation of the posterior tension band in this manner restores the normal anatomy after tumor resection, may promote bony fusion, and minimizes the potential for spinal deformity (Houten and Weiner 2000; Houten and Cooper 2000; Raimondi et al. 1976); (Constantini et al. 1996, 2000).
Several risk factors for development of a progressive spinal deformity have been identified, including preoperative scoliotic deformity, an increasing number of resections, an age less than 13 years, tumor-associated syrinx, surgery involving more than four levels, surgery spanning the thoracolumbar junction, and adjuvant radiotherapy (Yao et al. 2007; Ahmed et al. 2014a; Knafo et al. 2014; McGirt et al. 2008c). For multilevel surgery, data suggests that in situ fusion can decrease the risk of postresection deformity by 30 % and as much as 42 % in skeletally mature children (Anakwenze et al. 2011). Intraoperative localizing x-rays are crucial to identify the correct level of surgery. Once the dura is exposed, intraoperative ultrasonography improves the accuracy of surgical exposure and identification of the intramedullary tumor, which in turn reduces the size of the dural opening and myelotomy (Epstein et al. 1993; Maiuri et al. 2000; Hanbali et al. 2002; Brunberg et al. 1991; Raghavendra et al. 1984).
During tumor resection, real-time neurophysiologic monitoring is critical adjunct. Common modalities of monitoring include motor evoked potentials (MEPs), somatosensory evoked potentials (SSEPs), and measurement of D-waves (Nash et al. 1977; Morota et al. 1997; Goh et al. 2000; Calancie et al. 1998; Jones et al. 1996; Pechstein et al. 1996; Costa et al. 2013). Intraoperative changes in these signals can predict postoperative deficits (Quinones-Hinojosa et al. 2005; Cheng et al. 2014b). Neurophysiology can also be useful in delineating an appropriate entry point by mapping the dorsal surface of the spinal cord (Auguste and Gupta 2006; Cheng et al. 2014b). A bipolar stimulator can be swept from a lateral to medial direction until no SSEPs are recorded. This is then delineated as “septum,” and the process is confirmed from the contralateral side. This is especially helpful in cases where the tumor does not extend to the cord surface or if the anatomy is rotated or distorted.
The technique of tumor removal is determined by the surgical objective, tumor size, and gross and histological characteristics of the tumor. If no physical plane is present between the tumor and surrounding spinal cord, then it is likely that an infiltrative tumor is present. A biopsy is performed to establish a histological diagnosis. If an infiltrating or malignant astrocytoma is identified and is consistent with the intraoperative findings, further tumor removal may not be warranted. If tumor is easily identified, then continued removal is reasonable with close attention paid to motor and sensory evoked potentials. A reduction in these signals can predict postoperative deficits (Asazuma et al. 1999; Quinones-Hinojosa et al. 2005; Cheng et al. 2014a). Uncertainty of spinal cord–tumor interface should signal an end to tumor resection (Asazuma et al. 1999). On the other hand, ependymomas appear with a smooth, reddish-gray glistening tumor surface, which is sharply demarcated from the surrounding spinal cord. Large tumors may require internal decompression with an ultrasonic aspirator or laser, and the surgical goal in these cases is gross total resection.
10.6.1.3 Postoperative Management
Postoperatively, early mobilization is encouraged to prevent complications of recumbency such as deep venous thrombosis and pneumonia (Smith et al. 2004). Patients with severe motor deficits are particularly vulnerable to thromboembolic complications. Compression stockings are routinely used, and subcutaneous heparin (Epstein 2005) is begun on the second postoperative day in these patients. Orthostatic hypotension may occasionally occur following removal of upper thoracic and cervical intramedullary neoplasms. This is usually a self-limited problem that can be managed with liberalization of fluids and more gradual mobilization. A posterior fossa syndrome occasionally occurs following removal of a high cervical intramedullary neoplasm. Neck pain and stiffness can be managed with steroids and anti-inflammatory medications, although a lumbar puncture may sometimes be required to exclude a diagnosis of meningitis (Cooper and Epstein 1985; McCormick and Stein 1990). Early and aggressive use of physical and occupational therapy results in a better functional recovery.
Despite evidence to support a GTR, there is a risk of recurrence (Whitaker et al. 1991; Chang et al. 2002). Long-term clinical and radiographic follow-up is warranted in these patients (Sandalcioglu et al. 2005). An early postoperative MRI establishes the completeness of resection and serves as a baseline against which further studies can be compared. GTR is defined as more than 90 % tumor removal, subtotal resection (STR) as 50–90 %, and partial as less than 50 %. Serial gadolinium-enhanced MRIs are obtained because radiographic tumor recurrence usually precedes clinical symptoms (Chang et al. 2002; Hanbali et al. 2002). Serial radiographs should be obtained in high-risk patients to monitor for development of a progressive kyphotic deformity.
10.6.2 Radiation
Radiation therapy plays an adjunctive role in the treatment of malignant tumors and incompletely resected low-grade astrocytomas (Isaacson 2000; Guss et al. 2013). Low-grade astrocytomas and ependymomas that undergo GTR can be followed with serial imaging only. Radiotherapy in young children is associated with significant adverse effects, and therefore it is preferable to avoid or delay radiation therapy as long as possible in low-grade tumors (Rousseau et al. 1994; Perilongo et al. 1997; Prados et al. 1997; Gornet et al. 1999; Zuccaro et al. 1999; Grill et al. 2001; Teo et al. 2003; Valera et al. 2003).
GTR of grade II intramedullary ependymomas provides better long-term tumor control compared to STR and radiation therapy (McCormick et al. 1990b; Epstein et al. 1993; Hulshof et al. 1993; Cristante and Herrmann 1994; Hoshimaru et al. 1999; Lee et al. 2013). Although some authors recommend that radiation therapy is unnecessary following gross total resection (Cooper and Epstein 1985; Cooper 1989; McCormick et al. 1990b; Epstein et al. 1993; Hulshof et al. 1993; Samii and Klekamp 1994; Isaacson 2000; Kothbauer 2007), some studies have reported a 5–10 % recurrence rate following surgery (Guidetti et al. 1981; Cooper 1989; Hulshof et al. 1993; Chang et al. 2002). Surgery followed by external beam radiotherapy has been shown to result in 84 % local control of tumor for IMSCT of multiple tumor types (O’Sullivan et al. 1994).
Subtotal resection, as expected, has a very high recurrence rate (Cooper 1989; Linstadt et al. 1989; Chang et al. 2002). The data supporting postoperative radiation after STR is largely based on studies with small patient populations, limited follow-up, and inadequate controls treated without radiation therapy (Isaacson 2000; O’Sullivan et al. 1994). Despite these limitations, the overall results suggest that radiation may be beneficial after STR of spinal cord ependymomas (Kopelson and Linggood 1982; Garcia 1985; Shaw et al. 1986; Cooper 1989; Linstadt et al. 1989; Guss et al. 2013). The usual dose delivered is approximately 5,000 cGy in 180–200 cGy fractions using external beam radiation therapy. In some cases, reoperation and another attempt at GTR should be considered if a recurrent tumor is more accessible or better defined from the normal spinal cord (Cooper 1989; Chamberlain 2002b; Hanbali et al. 2002).
Patients who present with focal disease usually recur locally and do not manifest late dissemination (Chamberlain 2002a, b). Craniospinal radiation is only indicated for the rare patient who presents with multifocal disease (Garrett and Simpson 1983; Linstadt et al. 1989; Hulshof et al. 1993). Although the outcome is worse for this subgroup, good control rates have been reported (Garcia 1985; Linstadt et al. 1989).
10.6.3 Chemotherapy
Currently, chemotherapy is not routinely used in the initial treatment of IMSCTs, although some clinicians consider the use of chemotherapy for recurrence or in combination with radiation therapy for high-grade or incompletely resected tumors. Very little published data exists to provide insight into the impact of chemotherapy on outcomes, and chemotherapy regimens are typically based on those with some demonstrated activity in the setting of intracranial disease. Objective radiographic responses for supratentorial ependymoma have been observed with the use of combination platinum agent with etoposide, but the effect of chemotherapy on survival and functional outcome is still unclear (Massimino et al. 2002; Valera et al. 2003). The prospect of preoperative chemotherapy for second-look surgery has been explored in small trials with mixed results (Foreman et al. 1996; Schiffer and Giordana 1998; Chamberlain 2001; Valera et al. 2003). Also, because of the desire to avoid or delay radiation therapy in young children under 3 years of age, adjuvant chemotherapy may have a potential role (Prados et al. 1997). In a series of adult patients with recurrent spinal low-grade astrocytoma treated with temozolomide, there was a modest effect (Chamberlain 2008).
Chemotherapy guidelines for pediatric IMSCTs have been mainly derived from the clinical experience with intracranial ependymomas and low-grade astrocytomas. No randomized clinical trials have been performed (Kothbauer 2007). Etoposide, a topoisomerase II inhibitor, has been used to treat recurrent intramedullary ependymomas. This drug appeared to be well tolerated with modest toxicity (Chamberlain 2002a, b). Further trials are needed to determine the efficacy of this potential therapy for recurrent and refractory intramedullary ependymomas. Fakhreddine and colleagues demonstrated an association between chemotherapy (primarily temozolomide) and improved progression-free survival, but not overall survival, in infiltrative astrocytomas (Fakhreddine et al. 2013). They saw no association between extent of resection or adjunctive radiotherapy and outcome in these patients (Fakhreddine et al. 2013).
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


