Chapter 24 Inhibiting the Entry of R5 and X4 HIV-1 Phenotypic Variants
Compounds that prevent HIV-1 from entering its target cells are now available as a new therapeutic drug class, the entry inhibitors. They could play an important role in treating HIV-1 infection in future years, and they may also be valuable for preventing transmission, as preexposure or postexposure prophylaxis and/or as a topical microbicides. This chapter will review how the entry inhibitors might be used in clinical practice.
What are these compounds, how do they act, and what aspects of HIV-1 virology and pathogenesis may influence their use clinically? In addressing these issues, emphasis should be placed on the HIV-1 phenotypic variants known as R5 (CCR5-using) and X4 (CXCR4-using) viruses, because of their implications for the design of new therapeutic strategies in general, and those based on inhibition of viral entry via CCR5 and/or CXCR4 in particular. Most transmitted strains of HIV-1 use the CCR5 co-receptor for entry into CD4+ cells. Left untreated, infection with these R5 viruses is usually fatal. The X4 or R5X4 (CCR5- and CXCR4-using) strains can instead use the CXCR4 co-receptor and are rarely transmitted, (more accurately, they rarely dominate plasma viremia in the early years of infection), but when they do become relatively prevalent they are associated with an increased rate of disease progression. This is manifested by an acceleration in the rate of CD4+ T-cell loss, particularly of naive T cells, and a reduction in the regenerative capacity of the immune system. Hence the CXCR4-using strains represent a more lethal form of HIV-1.1–3
As described below, there are multiple stages to the process by which HIV-1 attaches to and fuses with target cells, the different stages being susceptible to intervention by different compounds that have different mechanisms of action. Some compounds inhibit virus-cell attachment, others interfere with the subsequent co-receptor interactions that drive fusion, yet more impede the later stages of the fusion process itself.1,4–7 Hence there has been a tendency to designate individual compounds as “attachment inhibitors”, “co-receptor inhibitors”, or “fusion inhibitors”. To some extent, the distinction is artificial, because all the compounds inhibit the entry process, albeit in different ways (Fig. 24-1). In this chapter, the generic term “entry inhibitors” will be used to describe all compounds that prevent HIV-1 from gaining access to the interior of the target cell. However, this chapter will focus only on compounds that bind specifically to the CCR5 or CXCR4 co-receptors. Inhibitors such as the BMS-378806 family of compounds that block gp120–CD4 attachment (see Chapter 27),8 and enfuvirtide (see Chapter 23), which prevents conformational changes in gp41 at a late stage of fusion,7 will be reviewed in detail elsewhere in this book. Note that the BMS-378806 family and enfuvirtide are active against both R5 and X4 viruses.
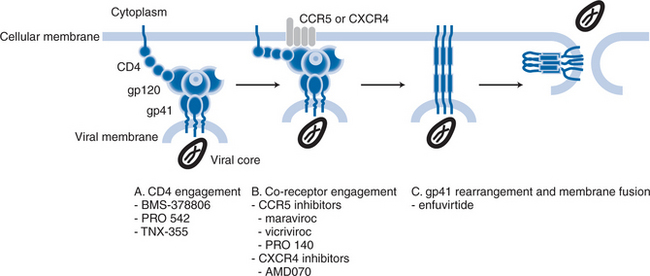
There is a semantic issue relating to co-receptor usage by the phenotypic variants. In the original nomenclature devised after the identification of CXCR4 and CCR5 as the principal co-receptors for HIV-1, isolates that use CCR5 were termed R5, while those using CXCR4 were called X4. Viruses that can use either co-receptor were designated R5X4 and they are often referred to as “dual-tropic”.9 Over the past decade, however, it has been appreciated that many uncloned “dual-tropic” isolates contain a mixture of R5 and X4 viruses, rather than individual viruses that are capable of entering cells via either co-receptor.3 A more accurate term for phenotypically mixed isolates is R5+ X4, with the term R5X4 being reserved for clonal viruses that can use both CCR5 and CXCR4 at the molecular level. Because the most commonly used commercial phenotyping assay (Trofile, from Monogram Biosciences, Inc) cannot discriminate between dual-tropic clonal viruses and phenotypic mixtures, an alternative terminology has arisen to describe samples that register as positive for entry into both CCR5-expressing and CXCR4-expressing cells: “Dual/Mixed” or D/M.10 We will not be using this terminology; instead, we will use the term R5+ X4 except when making specific reference to a single virus that is known to be able to use both co-receptors (R5X4).
THE HIV-1 ENTRY PROCESS IS MEDIATED BY THE VIRAL ENVELOPE GLYCOPROTEINS
HIV-1 fuses with target cells after a series of events that are triggered by binding of the viral envelope glycoproteins to specific receptors on the cell surface.11 The process of virus-cell attachment is usually the rate-limiting step for HIV-1 infection, just as it is for other viruses, because the negatively charged surfaces of both virions and cells create repulsive forces.12 In vitro, such effects can often be overcome by experimental devices such as the use of polycations to negate the charge imbalance, or by centrifugation of virions onto the cells.13 In the real world, however, HIV-1 uses ancillary receptors to facilitate its binding to the cell surface.12,14,15 Several such interactions have been described, their relative importance usually being cell type-dependent.12,14,15 Some ancillary interactions are mediated by the viral envelope glycoproteins, others by host cell-derived proteins or lipids that become incorporated into the viral membrane during egress of the virion from the cell.12,14,15 Examples of the former type of interaction include the charge-based binding of cationic patches on the envelope glycoproteins to anionic moieties on cell surface heparan sulfate proteoglycans, and the binding of envelope glycoprotein (Env) glycans to cell surface mannose C-type lectin receptors including, but not limited to, DC-SIGN.12,14 Alternatively, intracellular adhesion molecules (ICAM) incorporated into the virion membrane can associate with cell surface leukocyte function associated antigen-1 (LFA-1) to facilitate virus-cell attachment, particularly for the memory cells that are the preferred targets for R5 viruses.12,15
The ancillary interactions are neither necessary nor sufficient for triggering the events that lead to virus-cell fusion; they serve to concentrate virions on the cell surface in proximity to the fusion receptors proper, which increases the efficiency and rate of fusion, overall.12 Irrespective of how HIV-1 attaches to cells, the fusion-related events are initiated by the sequential interaction of the viral Env complex with CD4 and a co-receptor.11 The functional Env complex is a hetero-trimer of three gp120 surface glycoproteins, each noncovalently attached to three gp41 transmembrane glycoprotein subunits.16–20 There may be as few as 10 Env complexes on a typical HIV-1 virion,21 with perhaps only a single one being sufficient to mediate infection.22 The crystal structures of the CD4-bound and free forms of gp120, together with sophisticated models of the gp120 monomer and trimer, have greatly helped an understanding of how the Env complex interacts with its receptors.20,23–25 The structure of gp120 has been reviewed extensively elsewhere,26,27 but in summary, gp120 contains a gp41-binding surface comprising residues in the C1 and C5 regions; a CD4-binding site (CD4bs) formed from multiple, well-conserved residues folded into proximity; a co-receptor binding site formed from elements of the V3 region and/or other, more conserved structures comprised of elements of the bridging sheet; the sequence-variable V1V2 loop structure that overlies the more conserved receptor binding sites; and 20–35 N-linked (and perhaps also several O-linked) glycan residues that are located both in the five variable regions (V1–V5) and on more conserved sites and which serve to shield the protein surface from immune recognition.20,23–25 A key element of the CD4bs is a deep cavity that forms a contact site for an amino acid, Phe-43, protruding from a loop on CD4.25 Due to the conformational flexibility of gp120, this cavity is probably only transiently formed in the absence of CD4 and is stabilized by CD4 engagement.23 The binding of CD4 results in large conformational changes in other regions of gp120, including the formation of the co-receptor binding site.23,28 BMS-378806 and related compounds inhibit the gp120–CD4 interaction, and also subsequent conformational changes involved in the formation of the co-receptor binding site.8,29 These compounds are thought to intercalate within a cavity present in the CD4-free conformation of gp120, thereby stabilizing this conformation and inhibiting CD4 binding.23
The successful engagement of CD4 induces structural changes within gp120 that create, or expose, a binding site for the co-receptor.23 The nature of the binding site on gp120 is similar for each co-receptor, but critical, and as yet ill-defined, structural differences underlie which of the two co-receptors is actually used by any given virus.24,30,31 These changes in structure are usually created by sequence alterations in V3 that directly or indirectly affect the co-receptor-binding site.32 The complexities of the co-receptor-binding interactions and the role of the V3 region in influencing HIV-1 phenotype have been reviewed extensively elsewhere.32 In general, the V3 region of X4 viruses has a higher positive charge than the corresponding region of R5 viruses.32 Changes at only a few, specific residues in V3 can sometimes be sufficient to convert an R5 virus into an X4 variant,32 but additional changes elsewhere in gp120 are usually required to make a fully replication competent X4 virus.33
The engagement of the co-receptor causes structural changes in the gp41 subunits that drive membrane fusion.1,4,5,11,34 The gp41 protein contains two segments known as the heptad-repeat (HR) regions, HR1 and HR2, which have a locally α-helical structure; the three HR1 regions of the three individual gp41 subunits form a coiled-coil structure.6 During fusion, the interaction of HR1 with HR2 is considered to be the critical event that drags the viral and cell membranes toward one another.6 Thus, following insertion of the gp41 fusion peptide into the host-cell membrane, the HR2 region folds back into grooves on the coiled surface of the HR1 coil to form a structure that is commonly called, and depicted as, a hairpin.6 It is the formation of this highly stable structure, also often called the six-helix bundle, that releases enough stored energy to bring the viral and cell membranes close enough together for membrane fusion to occur.6 Enfuvirtide, a soluble mimetic of the HR2 structure, inhibits the intermolecular association between the HR1 and HR2 domains of gp41, to sterically prevent the conformational changes needed to form the six-helix bundle.6 Hence, fusion is arrested after the co-receptor-binding stage of the process (Fig. 24-1).
HIV-1 TROPISM: THE R5 AND X4 PHENOTYPIC VARIANTS
CCR5 and CXCR4 are the co-receptors most relevant to HIV-1replication in vivo. Although at least 10 other G-protein coupled receptors (GPCRs) can mediate entry of some HIV-1strains when they are expressed in transfected cells in vitro,35,36 the use of these receptors for entry into primary CD4+ cells is rare.37–48 Thus, the replication of HIV-1 in primary cells in vitro is almost always fully sensitive to inhibitors specific for CCR5 or CXCR4.46–48 In vivo, individuals who lack CCR5 expression are strongly protected against HIV-1 infection.49–51 On the rare occasions when CCR5-negative individuals become HIV-1 infected, their isolates use CXCR4 (and sometimes CCR5) but not other GPCRs to infect primary cells in vitro.52–55 In general, usage of GPCRs other than CCR5 or CXCR4 does not correlate with disease progression.40 Drug development programs aimed at blocking co-receptor binding have therefore focused only on CCR5 and CXCR4.5,56
Because patients can harbor HIV-1 tropism variants using either or both of the principal co-receptors, it is important to take these variants into account when making treatment decisions involving co-receptor inhibitors.3,57 The differential replication of R5 and X4 viruses in cell types that express CCR5 and CXCR4 differently is central to an understanding of HIV-1 pathogenesis.3,57 Indeed, as suggested, “HIV-1 infection of humans might almost be viewed as two separate diseases caused by two different lentiviruses that have different cellular tropisms: R5 HIV-1 strains, and their close cousins, the X4 viruses.”3
It was recognized almost 20 years ago that two different HIV-1 tropism variants existed.58–62 Isolates that formed syncytia in human peripheral blood mononuclear cells (PBMCs) were designated as syncytium inducing (SI), those that did not as NSI.62 Viruses able to replicate in primary human monocyte-derived macrophages (MDM) were called M-tropic, those in CD4+ T-cell lines, T-tropic.63,64 The formation of syncytia in MT-2 cells correlates well with the SI and T-tropic phenotypes, failure to do so with the NSI and M-tropic phenotypes.65,66 The MT-2 syncytium formation assay soon became the accepted standard for routine determinations of tropism.67–76 When CCR5 and CXCR4 were discovered, their cellular expression patterns explained tropism variants at the molecular level: CCR5 is the co-receptor for NSI/M-tropic viruses, CXCR4 for the SI/T-tropic strains.39,77 For these reasons, the nomenclature system based on the R5 and X4 terminology was designed (as discussed earlier).9
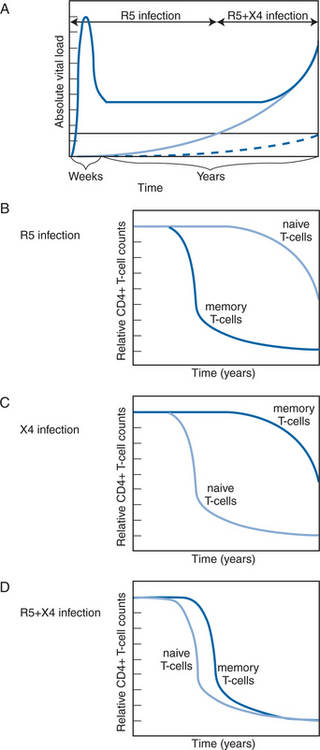
It is essential to recognize that R5 and X4 viruses do not infect all CD4+ T cells with equal efficiency. CXCR4 is expressed on naive T cells from which CCR5 is largely absent, but, conversely, CCR5 expression is high on memory CD4+ T cells, and CXCR4 low.78,79 This pattern of co-receptor expression is important for understanding how R5 and X4 viruses destroy the immune system at different rates. In a simplified model, R5 strains eliminate the memory CD4+ T-cell pool, and of course they also destroy the naive cells after they become activated and then express CCR5.2,3,80,81 These events gradually erode and eventually completely destroy the T-cell mediated immune system. The targeting of naive T cells by X4 viruses has the same effect, but more rapidly; the loss of the naive T-cell pool prevents the body from responding to new pathogens (Fig. 24-2).2,3
The R5 and X4 viruses tend to dominate plasma viremia at different times after initial infection. Most isolates or clones from primary infection cases have the R5 phenotype with few X4.3,39,77 Nonetheless, X4 viruses certainly can be transmitted; the rare infections that do occur in individuals homozygous for the defective CCR5-Δ32 allele involve X4 viruses.52–55 However, the overall protection provided by CCR5-Δ32 homozygosity shows the difficulties faced by X4 viruses in establishing new infections.49–51 The early prevalence of R5 viruses may reflect preferential transmission, but the same initial R5 dominance occurs irrespective of whether infection occurs sexually, vertically or via intravenous drug use.3,82 Hence a compelling alternative hypothesis is that R5 viruses are more efficiently amplified soon after transmission.3 If so, the initial target cells for viral amplification must be the same for all infection routes or, if different cells are involved, they must all favor R5 virus replication.3
Additional selection processes in favor of R5 viruses may operate at the level of the genital or rectal mucosa during sexual transmission. One hypothesis is that virions are transported by dendritic cells to lymph nodes where they are passed to activated memory CD4+ T cells, which express more CCR5 than CXCR4 and preferentially amplify R5 viruses.3,83 Activated, CD4+ CCR5+ memory T cells are particularly prevalent in the gut-associated lymphoid tissue (GALT) where they normally combat food-borne pathogens.81,84 The GALT therefore efficiently replicates R5 viruses during primary infection both in the macaque85 and humans.86,87 In both species, CD4+ CCR5+ T cells are substantially depleted from the GALT before this occurs in the peripheral blood, because cells of the GALT do not rapidly transit into the blood, and vice versa.85,88 The preponderance of R5 viruses in GALT could, however, reflect earlier selection pressures; the GALT could be seeded with R5 viruses that had been preferentially transmitted and amplified in draining lymph nodes.89 The rapid and selective elimination of activated memory CD4+ CCR5+ T cells from GALT by R5 viruses may suggest that tropism variants selectively deplete the subsets of CD4+ cells that express the appropriate co-receptor.2,3,80,81 However, considerable complexities and controversies surround this issue, and direct cell killing is unlikely to be sufficient to explain viral pathogenesis, particularly for R5 viruses.2
Although they are rare early in infection, X4 viruses eventually become detectable in the blood of ∼50% of individuals infected with subtype B viruses.72,77,90–93 The appearance of X4 viruses at levels detectable by phenotypic assays typically takes ∼5 years, but it can take much less time.72,77,90–93 Death from AIDS does not require the appearance of X4 viruses at detectable levels in the blood, but the emergence of X4 viruses in both acute and chronic infection is associated with accelerated CD4+ T-cell count decline, and the relatively rapid onset of AIDS and death.67–71,73–76,94 Even in chronic infection, however, it is unusual for X4 viruses to dominate plasma viremia; in most cases, the infection is phenotypically mixed (R5+ X4), with the R5 component being the major contributor to the total viral load.92,94
It is still uncertain whether the emergence of X4 viruses is the cause or consequence of immune destruction; the interrelations between the viral phenotype and the immune system can be complex.3 The preferential tropism of X4 and R5 viruses for naive and memory cells, respectively, has been used to model the tropism switch under in vivo conditions.95 Memory T-cells divide ∼10 times as often as naive cells in uninfected people. Because HIV-1 replication is more efficient in CD4+ T cells that are activated, the tropism of R5 viruses for memory cells could provide them with a quantitative advantage in early infection.96,97 In lymphoid tissue blocks, in vitro, ∼10-fold more R5 viruses than X4 viruses are produced on a per cell basis; their “burst-size” is higher.98 However, during disease progression, and at low CD4 counts in particular, the division frequency of naive T cells approaches that of memory cells.99,100 Since naive T cells are more abundant in vivo, particularly after R5 viruses have depleted the memory T-cell pool, this scenario provides a quantitative basis for the X4 switch late in disease.95 Low CD4 counts may therefore both drive and arise from the late dominance of X4 viruses.95 Such arguments underline the old cause-or-consequence controversy and imply that the R5 to X4 switch process might involve a lethal spiral.95 Overall, R5 viruses are deadly, their X4 cousins deadlier.57
The tropism variants destroy the immune system in different ways, something clearly demonstrated in the macaque model. A pathogenic R5 SIV gradually, but eventually completely, depletes the memory CD4+ T-cell pool, whereas an X4 simian human immunodeficiency virus (SHIV) very rapidly destroys the entire repertoire of naive T cells in the periphery and lymphoid tissues.80,101,102 Indeed, X4 SHIVs cause ultra-rapid progression to AIDS; the extremely rapid loss of CD4+ T-cell help for B cells even prevents the animals from developing antibody responses to viral antigens.103 Rapid progression can also occur in a small minority of macaques infected with an R5 SIV, emphasizing the importance of host variation as an influence on the rate of disease.104–106
Although most R5 viruses replicate in macrophages, the R5 phenotype and macrophage tropism correlate imperfectly.107 For example, some X4 viruses can replicate in macrophages, for reasons that probably lie in considerations of CD4+, CCR5 and CXCR4 density which are much lower on macrophages than on activated CD4+ T cells.54,108–112 We noted earlier that R5 viruses are pathogenic even when X4 strains cannot be detected. An increased ability to replicate in macrophages is one correlate of disease progression mediated by R5 viruses.113,114 However, it is unclear whether this is a direct consequence of HIV-1 replication in macrophages or a surrogate marker for a viral adaptation to use low CD4 and CCR5 levels more efficiently.115 Thus, any remaining CD4+ T-cells expressing only low levels of CCR5 would also become vulnerable to more virulent R5 variants with an increased CCR5 affinity. Any increase in the ability of HIV-1 to use low CCR5 levels via an affinity increase might result in decreased susceptibility to CCR5 inhibitors and other entry inhibitors.115,116 This point will be revisited below, because it is relevant to a consideration of when is the best time to start therapy with CCR5 inhibitors; their early use may be favored on virological grounds, as well as immunological ones, although safety issues must also be considered.57
Although most plasma virus is derived from CD4+ T cells, infected macrophages can sustain very high levels of virus replication.106,117,118 Thus, in macaques infected with X4 SHIVs all the naive CD4+ T cells are rapidly eliminated to the extent that no CD4+ T cells can be detected in the peripheral blood or lymphoid tissues.80,101,102 Yet plasma viremia levels remain extremely high because X4 viruses are produced by infected macrophages.117,118 These cells may also be responsible for producing the even higher levels of R5 SIV that are sustained in dual-infected macaques after depletion of the CD4+ T-cell pool by an X4 SHIV.106,119 Hence it will be useful to determine what effects CCR5 and CXCR4 inhibitors have on macrophage infection in vivo.
Some central questions about HIV-1 co-receptor use in vivo remain unanswered. For example, it is still not clear why X4 viruses emerge (or re-emerge) at high levels after several years only in some individuals and not in others. What host factors are the most relevant here? For more detailed considerations of the relationships between HIV-1 tropism and the immune system, additional reviews should be consulted.2,3,81,84,107,120,121
ASSAYS TO DETERMINE HIV-1 TROPISM
Given the importance of tropism as an influence on HIV-1 pathogenesis, and conceivably on treatment options, it follows that there should be accurate methods for the routine identification and quantification of tropism variants. The MT-2 syncytium formation assay indirectly measures co-receptor use because MT-2 cells express CXCR4 and CD4, but not CCR5.3 Thus, if an isolate forms syncytia in MT-2 cells, it contains X4 or R5X4 viruses. More recently, assays using recombinant viruses have now developed to assess the co-receptor usage of clinical isolates. The Phenoscript (VIRalliance, Paris, France) and Trofile (Monogram Biosciences, Inc, South San Francisco, CA, USA) assays are available commercially.10,122 HIV-1 env sequences are amplified in bulk from plasma and used to produce replication-competent viruses (Phenoscript) or replication-defective pseudoviruses (Trofile).10,122 These viruses are used to infect target cell lines engineered to express CD4 and either CCR5 or CXCR4, the outcome being determined by activation of a reporter gene present in the cells (Phenoscript) or in the incoming viruses (Trofile).10,122 As noted above, when a plasma sample registers as positive on both CCR5- and CXCR4-expressing cells, it usually means it contains a mixture of R5 and X4 viruses. A truly R5X4 virus cannot be distinguished from a phenotypic mixture in assays such as Phenoscript or Trofile.3,123
There are limitations to the present generation of tropism assays and since they could affect treatment decisions, they need to be understood. The most troublesome problem is, perhaps, the detection threshold for viruses of one phenotype in the presence of a larger amount of the other one.57 For example, the Trofile assay can only reliably detect X4 viruses in mixtures when they constitute at least 10% of the total.10 As an example of why this limit could cause relevant virological changes to go undetected, in a macaque that was deliberately dual-infected with an R5 SIV and an X4 SHIV the X4 component constituted ∼4% of the total viral load at baseline, as measured using a highly specific, PCR-based discriminatory viral load assay.119 It is questionable whether this animal would have registered as dual-infected using the Trofile assay, and whether that assay could have properly quantified the transient increase then decrease in the X4 component that occurred during CCR5 inhibitor therapy.119
The absolute sensitivity limit of the Trofile assay, i.e., the lowest viral load at which it can reliably detect the presence of viruses of either phenotype, is also an issue.10,122 Such considerations are important, because very low plasma levels of X4 viruses may still have clinical significance, even if they cannot be readily detected and quantified.92 An example is the accidental recruitment of a patient into a phase I trial of a CCR5 inhibitor who harbored low levels of X4 viruses; as per protocol such individuals were excluded from this study, but the screening assay (Trofile) was initially unable to detect the X4 component of the viral load.124 In clinical practice, it would also be very important to discriminate between an absolute increase in the amount of X4 viruses during CCR5 inhibitor therapy (see further ahead), and a relative increase caused because the R5 component of a mixed R5+ X4 infection is lowered by the therapy (Fig. 24-3).57 The latter outcome would not necessarily be a bad one, but any sustained switching of co-receptor usage would certainly be unwanted. A truly useful phenotypic assay must, therefore, be able to discriminate satisfactorily between the above scenarios.
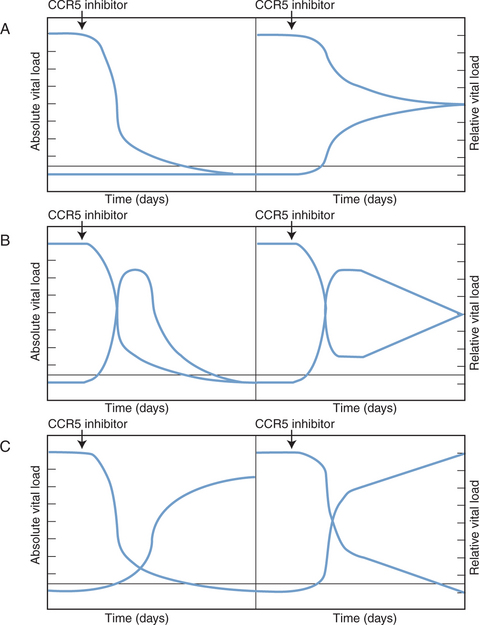
Figure 24-3 Theoretical scenarios for the effects of CCR5 inhibitor therapy on R5 and X4 virus loads. In each scenario, the left panel depicts absolute viral loads (in arbitrary units), with R5 viruses high and X4 viruses undetectable. The right panel shows relative viral loads (i.e., the relative contributions of R5 and X4 viruses to the total viral load). The threshold for detection of X4 viruses in a phenotypic assay is indicated by the horizontal line. (A) The CCR5 inhibitor selectively blocks R5 virus replication and has no effect on X4 viruses. The absolute level of X4 viruses is unaffected, but because R5 viral load drops, the relative contribution of X4 viruses to the total viral load increases. In theory, this might make X4 viruses become detectable in phenotypic assays. Whether this could happen in practice is an important issue to resolve. (B) Transient increases in both absolute and relative X4 viral loads occur when the R5 viral load declines as a result of CCR5 inhibitor therapy. This scenario is similar to what was seen in two of three R5+ X4 virus infected macaques treated with the R5 inhibitor CMPD 167.119 Whether it would happen in HIV-1 infected people remains to be determined. (C) In this scenario, the therapy-induced decline in R5 viral load causes a sustained, possibly reciprocal, increase in X4 viral load. This is not a consequence of viral escape (i.e., R5 to X4 interconversion) but is due to expansion of X4 viruses into a niche vacated by R5 viruses.3 Again, whether this will occur in clinical practice is unknown. In all three scenarios the initial X4 viral load is depicted as being below the threshold of detection in a phenotypic assay because CCR5 inhibitors are most likely to be tested clinically under these conditions. If the phenotypic assays are sensitive to changes in absolute viral load, as depicted on the left of each panel, then it should be possible to resolve between the different scenarios. However, if relative viral load has a significant influence on the clinical performance of these assays, as depicted on the right of each panel, it may not be easy to determine which scenario most applies to the clinical data. This would complicate the taking of clinical decisions. In practice, CCR5 inhibitors will not be tested as monotherapy, but we have not attempted to model the influence of other drugs that are active against both R5 and X4 viruses.
Reproduced from Kuhmann SE, Moore JP. The HIV-1 phenotypic variants: deadly and deadlier. J Viral Entry 1:4, 2005.
The present generation of phenotypic assays use cell lines engineered to express CCR5 or CXCR4 in combination with CD4.10,122 Co-receptor usage on transfected cell lines and primary cells does not always correlate.47,125–127 For example, some viruses that score as R5X4 (or R5+ X4) on cell lines only use CXCR4 to enter primary CD4+ T cells.47,126,127 A similar discordance might affect the diagnosis of resistance to CCR5 inhibitors.128 Thus, CCR5 and CXCR4 adopt an array of conformational states on cell surfaces.129 If the relative amounts of different configurations vary from cell to cell, there could be a cell type-specific discordance in how resistant viruses enter different cell types. The expression levels of CCR5 and CXCR4 on primary and engineered cells also differ, another potential influence on the output of phenotypic assays.57
Significant efforts have also been applied to defining genetic algorithms that predict tropism, the basis of a genotypic assay for CCR5 and CXCR4 use.130–134 These methods are mostly based on analyzing the sequence of the V3 region of gp120, because a few ‘signature residues’ in V3 are correlated with the R5 and X4 phenotypes.132,134,135 The predictive powers of the genotyping assays are increasing, but gp120 sequence variation is sufficient to create imperfections in phenotype-genotype correlations.32,134 As an example of the plasticity of gp120, viruses completely resistant to small molecule CCR5 inhibitors can sometimes have sequence changes in V3, and sometimes not.128,136–138 Thus it may be very difficult to develop genetic assays of resistance to CCR5 inhibitors; signature residues for resistance may simply not exist, or their presence may be blurred by other changes within or outside V3. Additional improvements will be needed if genotypic assays are to be used routinely for co-receptor phenotype assessment in the clinic.132
Several population-based investigations of co-receptor tropism have been reported using the Trofile assay (Table 24-1). In general, treatment-naive populations with higher CD4 cell counts have the greatest proportion of R5 viruses; as the CD4 cell count falls, and also after the use of antiretroviral therapy, the prevalence of R5+ X4 viruses increases. In a cross-sectional assessment, co-receptor tropism could be successfully determined in 979 (82%) of 1191 HIV-1-infected, treatment-naive patients from the HOMER cohort in British Columbia: of these, 82% had R5 virus, 18% had R5+ X4 virus and only a single patient (<0.1%) had X4 virus.139 Patients for whom viral phenotypes could be determined had higher HIV-1 RNA levels (130 000 vs 61 000 copies/mL, P < 0.001), lower CD4 cell counts (260 vs 330/mm3, P = 0.007), and were less often CCR5-Δ32 heterozygotes (13% vs 20%, P = 0.007), compared to those that yielded no phenotypic information. Individuals harboring R5+ X4 viruses had higher HIV-1 RNA levels (175 000 vs 120 000 copies/mL, P = 0.006), lower CD4 cell counts (110 vs 290/mm3, P < 0.001), and more often had a prior AIDS diagnosis (22% vs 11%, P < 0.001) than patients with only R5 viruses. In a multivariate analysis, both a higher HIV-1 RNA level and a lower CD4 cell count were associated with the presence of an R5+ X4 virus. The association between R5+ X4 virus and a lower CD4 cell count was particularly strong; 54% of patients with CD4 cell counts <25/mm3 harbored an R5+ X4 virus but only 7% of patients with CD4 >500/mm3 did so.139
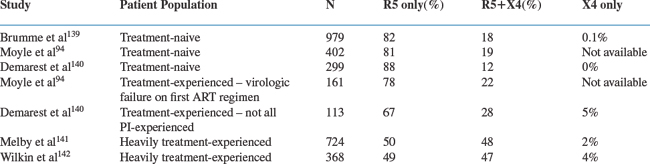
Another cross-sectional analysis was conducted on 861 patients followed at the Chelsea and Westminster Hospital in London.94 Viral phenotypes were successfully determined for 563 (65%) of the 861 patients tested (including 19% with nonsubtype B viruses, 65% who were treatment-naive patients and 35% who had failed their first antiretroviral regimen). Tropism results were more frequently obtained from patients with higher HIV RNA levels (39 325 vs 12 793 copies/mL, P < 0.001), but less often from those whose viruses were from outside subtype B (16% vs 20%). In the 402 treatment-naive patients with tropism results, 81% had R5 virus and 19% had R5+ X4 or X4 virus; in the 161 treatment-experienced patients failing their first antiretroviral regimen, 78% had R5 virus and 22% had R5+ X4 or X4 virus (P = 0.35). Overall, only four patients (0.7%) had exclusively X4 virus. Patients with R5+ X4 or X4 viruses had higher HIV-1 RNA levels (4.82 vs 4.55 log10 copies/mL) and lower CD4 cell counts (231 vs 307/mm3) than those with R5 viruses; no significant associations with antiretroviral treatment experience or viral genetic subtype were seen. In a multivariate analysis, the presence of an R5 virus was significantly associated with a lower HIV-1 RNA level, a higher CD4 cell count, and a lower natural killer (NK) cell count.94
A cross-sectional analysis of tropism was also conducted in 412 patients enrolled in three clinical trials of a CCR5 inhibitor.140 The group consisted of 299 treatment-naive patients and 113 patients taking a failing antiretroviral regimen. Among the treatment-naive patients, 88% had R5 viruses, the remaining 12% R5+ X4 viruses, while in the experienced patients, 67% were R5, 28% R5+ X4 and 5% X4. The presence of R5+ X4 viruses was associated with a decreasing CD4/CD8 ratio in treatment-naive subjects (P < 0.0001) and with increased age (P = 0.039), nonwhite ethnicity (P = 0.002) and prior protease inhibitor (PI) treatment (P = 0.009) in treatment-experienced patients.140
Other assessments of tropism have been conducted in heavily treatment-experienced patients. From the TORO studies of enfuvirtide, 724 three-class treatment-experienced patients underwent tropism testing at baseline, of whom 50% had R5 viruses, 48% R5+ X4 and 2% X4.141 Regression analysis identified an association between R5+ X4 virus and lower CD4 cell counts (P < 0.001), although R5+ X4 viruses were also identified in 40% of patients with CD4 cell counts >200/mm3. No association was seen between R5+ X4 virus and the number of antiretroviral drugs previously taken, the baseline phenotypic sensitivity score, or the prior use of the PI, lopinavir/ritonavir (all p > 0.5).141
In the AIDS Clinical Trials Group 5211 study, heavily treatment-experienced patients were screened for tropism at baseline with results obtained from 368 participants.142 Of these patients, 48% harbored R5 viruses, 48% R5+ X4 viruses, 4% X4 viruses. There was no influence of gender on tropism; 52% of women and 51% of men harbored R5 viruses. However, R5 viruses were more common in white (52%) and Hispanic (67%) patients than in blacks (42%) (P = 0.06). In multivariate analysis, the presence of an R5+ X4 or X4 virus was associated with a lower CD4 cell count (P = 0.03), but not with the HIV-1 RNA level or with drug resistance mutations, compared to when R5 viruses were present.142
Tropism was also determined for viruses isolated from both plasma and cerebrospinal fluid (CSF) in a cross-section of 46 patients from San Francisco, 80% of whom were treatment-naive, the remaining 20% treatment-experienced.123 Of the plasma viruses, 83% were R5, 17% R5+ X4, virtually identical to what was observed for the CSF viruses: 85% R5, 15% R5+ X4. Pure X4 viruses were present in none of the plasma or CSF samples. The tropism determinations were concordant for most patients. 78% had R5 viruses and 11% had R5+ X4 virus in both the plasma and CSF samples. However, tropism was discordant in five (11%) of the patients: two had R5 viruses in the plasma but R5+ X4 viruses in the CSF, whereas three had R5+ X4 viruses in the plasma and R5 viruses in the CSF. In principle, the discordant tropism observed in the CSF could have implications for CCR5 inhibitor therapy.123
THE BINDING OF GP120 TO CCR5 OR CXCR4 AS A DRUG TARGET
As noted earlier, the CD4-triggered binding of gp120 to a co-receptor is a critical stage in the fusion pathway, and it is the step that is interfered with by the class of HIV-1 drugs now referred to as co-receptor inhibitors (Table 24-2, Fig. 24-4).56,143–145
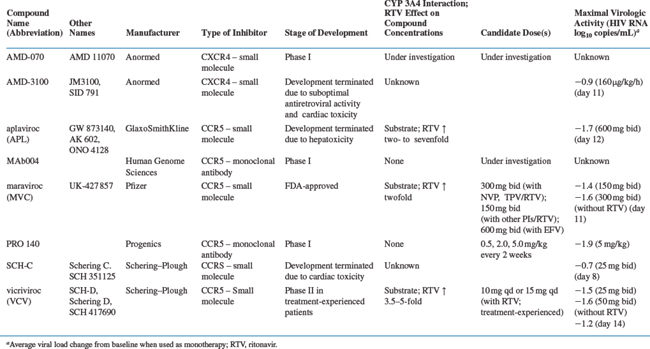
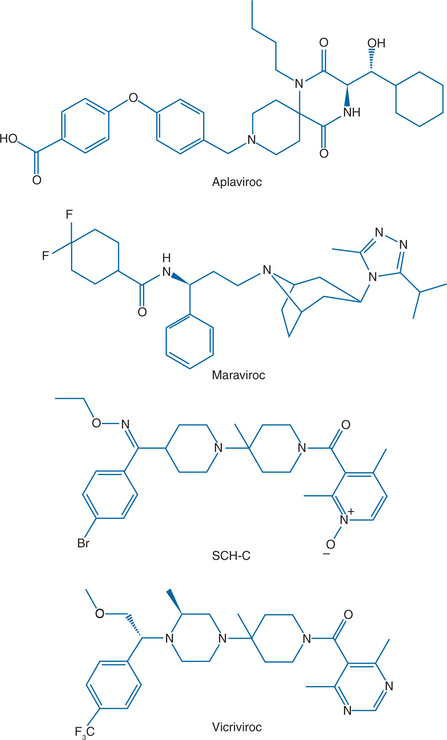
Figure 24-4 The structures of four CCR5 inhibitors that have shown antiviral activity in HIV-1-infected patients.
The binding of gp120 to CCR5 or CXCR4 involves multiple, discontinuous regions of the various proteins that form complex binding sites for one another. The co-receptor-binding site on gp120 is fully formed only after gp120 interacts with CD4.23 The V3 region and the conserved β19 strand are the most important gp120 regions involved in CCR5 and CXCR4 binding,24,30,31 the V3 residues contributing specificity for one co-receptor versus the other.32
Two distinct regions of CCR5 and CXCR4 interact with gp120, the N-terminus (Nt) and the second extracellular loop (ECL2).146,147 There are four tyrosine residues in the CCR5 Nt, each of which can be sulfated.148 Tyrosine sulfation, particularly at Tyr-10 and Tyr-14, is important for the efficient binding of gp120 to CCR5 or CXCR4, and viral entry, as well as for chemokine binding.149–153
The nature of the gp120–co-receptor interaction is probably broadly similar for CCR5 and CXCR4. Our current understanding is that the negatively charged co-receptor Nt probably associates with cationic gp120 residues in the β19 strand and near the V3 base, while ECL2 residues interact with the V3 crown.24,32,154 However, subtle differences also need to be taken into account. Thus, V3 residues may be more important for gp120 binding to CXCR4 than to CCR5,32 and the ECL2 region seems to play more of a role than the Nt in the gp120–CXCR4 interaction, whereas the converse may be true for gp120–CCR5 binding.147
The small molecule CCR5 inhibitors discussed below do not, however, directly block the gp120–CCR5 interaction. Instead, they act by inducing allosteric changes in CCR5 that render it unrecognizable by gp120.129,155–158 The same mechanism almost certainly operates for the CXCR4 small molecule inhibitors as well, although this has not been studied in any detail. Based on mutagenesis studies and molecular modeling, the putative binding site for several small molecule CCR5 inhibitors is a small cavity formed between the transmembrane helices on the extracellular face of CCR5 and distant from the gp120 binding site.156,159–161 Although it is likely that the details of how these inhibitors bind to this pocket differ from one compound to the next, their similar mechanisms of action imply that their binding sites are also broadly alike.157 The comparative studies that have been performed with the investigational CCR5 inhibitors AD101, SCH-C, and TAK-779 suggest that each compound has a very similar dependence on residues that line the inhibitor-binding pocket within transmembrane helices 1, 2, 3, and 7, albeit with some compound-specific variation.156,160,162 Mutational analyses of the binding sites for the investigational CCR5 inhibitors TAK-220 (Takeda) and CMPD 167 (Merck) indicate that they substantially overlap with those for AD101, SCH-C, and TAK-779, with additional contributions from residues in the other transmembrane helices, particularly helix 6.159,161 Similar studies with the FDA-approved CCR5 inhibitor maraviroc have also emphasized the subtle compound-specific differences in the way these antagonists interact with CCR5.163 In addition, some CCR5 amino acid residues may be important for the activity, but not the binding, of the small molecule inhibitors, lending support to the view that these compounds act allosterically and not competitively.155
Monoclonal antibodies (MAbs) such as PRO 140 are likely to act differently from the small molecules by directly impeding access of gp120 to its binding site on CCR5 inhibitor, although there are some uncertainties about the mechanism.164,165 The binding site for PRO 140 is probably identical to that of the murine MAb PA14 from which it was derived by humanization; it involves residues in the CCR5 Nt and ECL-2 and so essentially overlaps the binding site on CCR5 for gp120.147,165
Studies of how the investigational CXCR4 inhibitor AMD3100 binds to CXCR4 suggest that the binding pocket for this inhibitor also lies within the TM helices, facing the outside of the cell.166,167 The pocket is probably lined with several acidic residues that interact with positively charged atoms in AMD3100.166 How AMD11070 binds to CXCR4 has not been reported, but a reasonable supposition is that it interacts with the same pocket.
A notable feature of the small molecule CCR5 inhibitors is their long half-life on the receptor, which can exceed 1 week in a cell-free system.157 The longevity of the inhibitor-receptor complex may underlie delayed rebounds in plasma viremia seen after the cessation of therapy in both HIV-1-infected humans and in some SIV-infected macaques.168,169 There may also be a prolonged interaction between AMD3100 and CXCR4.170 It can reasonably be expected that PRO 140 and similar MAbs would also have a long receptor half-life as well. However, these various inhibitors do not downregulate CCR5 from the cell surface, they occupy it in a way that renders it unusable by HIV-1. Because the small molecules also prevent chemokines from binding and signaling, the disruption of a feedback loop can increase the local concentration of chemokines.171 The mechanism may be similar to that which elevates chemokine levels in cultures of CD4+ T cell from individuals with the CCR5-Δ32 allele.172 In principle, these chemokines could either reinforce the antiviral activity of the inhibitors or, alternatively, cause immunological complications.
THE CLINICAL DEVELOPMENT OF CCR5 INHIBITORS
Several orally available small molecule CCR5 inhibitors have been evaluated in HIV-1-infected people: aplaviroc (formerly GSK-873140) from GlaxoSmithKline, maraviroc (formerly UK-427 857) from Pfizer, and SCH-C and vicriviroc (formerly SCH-D), both from Schering Plough (Fig. 24-4).173–176 Antiviral efficacy was demonstrated for all four compounds, the mean viral load reduction at optimal dosing in 10–14 day monotherapy trials was in the range 1.5–1.7 log10.173–176 This reduction is comparable to what was observed with another small molecule CCR5 inhibitor, CMPD 167, in SIV-infected macaques.119,169 The SCH-C and aplaviroc clinical development programs have now been terminated, while maraviroc is FDA-approved and vicriviroc is in advanced clinical trials. Two humanized or human anti-CCR5 MAbs, PRO-140, from Progenics and CCR5mAb004 from Human Genome Sciences are now in phase I trials as injectable drugs.5,56,144,177 The clinical development of all these compounds is discussed in more detail below.
Aplaviroc
Aplaviroc (APL, formerly GW 873140, AK 602, and ONO 4128) is a small-molecule spirodiketopiperazine compound that entered clinical trials. It has potent antiretroviral activity in vitro with an IC50 <4 nM, including against viruses from multiple genetic subtypes.178 APL acts additively/synergistically in vitro with RTIs; PIs; enfuvirtide; other investigational CCR5 inhibitors; and investigational CXCR4 inhibitors179 (see further ahead for a discussion of “synergy”). Like all the CCR5 small molecules, APL is an allosteric, noncompetitive inhibitor that prevents gp120 binding.157 As noted above, the half-life of the inhibitor-CCR5 complex in a cell-free system is very long for all the small molecule drug candidates (>136 h for APL itself, slightly less for other tested compounds).157 This may have beneficial clinical consequences; the median CCR5 receptor occupancy in eight HIV-1-negative subjects following 7 days of APL administration was 98%, and in a clinical study of 31 HIV-1-infected subjects, the half-life of APL binding to CCR5 was 122 h.180 APL is readily absorbed orally and is a substrate for the hepatic cytochrome P450 (CYP450) 3A system.174 The area under the curve (AUC) concentration was dose-dependent, with some accumulation observed during a 10-day clinical dosing study.174,181
A phase I double-blind, randomized, placebo-controlled clinical study involved 40 HIV-1-infected individuals (19 treatment-naive and 21 treatment-experienced) with only R5 virus detectable (Trofile assay), a CD4 cell count nadir >200/mm3 and a plasma viral load >5000 RNA copies/mL who had not received antiretroviral therapy in the preceding 12 weeks. Study subjects were randomized to one of four dosing cohorts for 10 days: APL at 200 mg once or twice daily, 400 mg daily, 600 mg twice daily, or matching placebo. The baseline viral load was ∼4.36 log10 (23 000) RNA copies/mL, the CD4 cell count was ∼350/mm3.174
During the 10 days of APL dosing, there was a dose-dependent decrease in viral load, ranging from a mean change of −0.46 log copies/mL (at 200 mg daily) to −1.66 log copies/mL (at 600 mg twice daily) (Fig. 24-5). The nadir HIV-1 RNA level occurred at day 12 (i.e., 2 days after discontinuation of APL dosing). Viral load reductions were comparable in treatment-naive and -experienced subjects, and no changes in CD4 cell counts were observed. The most commonly reported drug related adverse events were gastrointestinal. They were diarrhea (25–37% APL, 0% placebo), loose stools (13–38% APL, 33% placebo), and nausea and abdominal pain (13–38% APL, 11% placebo). Gastrointestinal events occurred most commonly on study day 1 and were resolved within 1–3 days while therapy was continued. In one subject in the 200 mg daily dosing group, an R5+ X4 virus was detected by the Trofile assay on day 10 but only R5 viruses were present on day 24. The study investigators concluded that APL had potent antiretroviral activity and was generally well tolerated.174
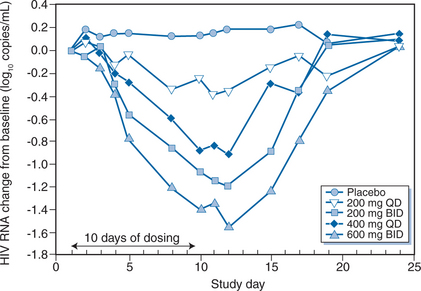
Figure 24-5 Mean change in log10 plasma viral load over time during treatment with aplaviroc by dose group.
Reproduced from Lalezari J, Thompson M, Kumar P, et al. Antiviral activity and safety of 873140, a novel CCR5 antagonist, during short-term monotherapy in HIV-infected adults. AIDS 19:1443, 2005.
The effects of APL on plasma chemokine and cytokine levels have also been assessed clinically.182 A total of 30 HIV-1-negative individuals received APL (200, 600, or 800 mg) or a placebo twice daily for 7 days, while 40 HIV-1-infected subjects were given the drug at 200 mg daily or twice daily, 400 mg daily or 600 mg twice daily, or received a placebo, for 10 days. In all participants, 10 of the 11 chemokines (RANTES, MIP-1α, IL-8 and MCP-1) or Th1/Th2 cytokines (TNF-α, IFN-γ, IL-2, IL-4, IL-5, IL-10) monitored were unaffected by APL administration. MIP-1β levels did increase transiently by 1.5–2.5-fold in the APL recipients, but remained within the normal range. The study investigators concluded that APL does not induce overt, global changes in the extent of immune activation.182
Drug–drug interactions can be anticipated with APL because it is a CYP450 3A substrate and such interactions have now been observed clinically. Thus, APL concentrations were increased by two- to threefold and by six- to sevenfold upon co-administration of ritonavir and lopinavir/ritonavir respectively, with no change in lopinavir levels and about a 30% increase in the ritonavir concentration.181 Similarly, co-administration of atazanavir/ritonavir and APL increased APL concentrations seven- to 17-fold without effect on atazanavir levels.183 Conversely, efavirenz co-administration decreased APL levels by 57% with no change in efavirenz concentrations.184 However, no such APL concentration decrease was seen with co-administration of efavirenz and fosamprenavir/ritonavir.185 Hence pharmacokinetic boosting with ritonavir could be useful to overcome the negative impact of efavirenz. No drug–drug interactions were observed when APL and tenofovir were co-administered.186
In September 2005, GlaxoSmithKline announced it was stopping its phase IIb dose-ranging studies of APL in treatment-naive patients, due to the occurrence of severe hepatotoxicity in a study participant.187 The patient was a 39-year-old man with no history of viral hepatitis and normal baseline hepatic transaminase and bilirubin levels who was randomized to receive APL 800 mg twice daily together with zidovudine/lamivudine.188 On study day 59, he developed severe hepatotoxicity; a liver biopsy revealed a chronic inflammatory infiltrate of moderate intensity, consistent with drug-induced hepatotoxicity. Upon discontinuation of the study medications, hepatic transaminase levels returned to baseline by study day 100. Further investigation revealed that among 282 subjects receiving APL in phase IIb studies, there were three additional cases of severe hepatotoxicity. In addition, one of 26 randomly assigned, treatment-experienced individuals in a phase III trial of APL had a severe elevation in hepatic transaminases at study week 4.188 This event led to the termination of the phase III trial, and further clinical development of APL was stopped in October 2005.189 In total, four (1%) APL recipients had ALT levels greater than three times the upper limit of normal (ULN) and bilirubin levels greater than 1.5 times the ULN, although none developed liver failure or died.188
Maraviroc
Maraviroc (MVC, formerly UK-427,857) is a small-molecule cyclohexanecarboxamide compound selected from ∼1000 imidazopyridine analogs that were screened for binding to CCR5.190 MVC has potent antiretroviral activity (mean IC90, 2 nM) against R5 viruses of diverse geographic origin and from multiple genetic subtypes. It is also active against isolates from both treatment-naive patients and those who had taken currently available antiretroviral drugs. As expected, MVC is inactive against X4 viruses. In vitro, MVC acts additively/synergistically with reverse transcriptase inhibitors (RTIs), PIs, and enfuvirtide. MVC binding to CCR5 inhibits the binding of MIP-1α, MIP-1β and RANTES, but does not itself trigger release of intracellular calcium; thus in common with the other small molecule CCR5 inhibitors, MVC is a chemokine antagonist but not a CCR5 agonist. MVC inhibits both HIV-1 gp120 binding to CCR5 (IC50, 11 nM) and HIV-1 entry into host cells (IC50, 0.22 nM).190 The dissociation half-life of MVC from the CCR5 receptor is ∼16 h.191
In animal studies, MVC was absorbed rapidly and was 40% orally bioavailable; in humans, it was 23% bioavailable and had a half-life of 13 h, allowing once or twice daily dosing.190,192,193 MVC is moderately lipophilic, but has relatively poor membrane permeability; its transcellular transport is mediated by the P-glycoprotein pathway. In oral dose escalation studies, MVC pharmacokinetics were nonlinear, leading to greater than expected concentrations at higher doses; thus, for a 10-fold increase in dose, Cmax increased 40-fold and AUC increased 20-fold. MVC is a substrate (but not an inducer or inhibitor) of the hepatic cytochrome P450 3A4 enzyme system, is metabolized by oxidation, and is excreted predominantly unchanged (72–94% in feces and 5–20% in urine). There was no evidence of MVC accumulation over 10 days of dosing.173
No cytotoxicity or adverse effects of MVC were identified in experiments using in vitro models of human immune system function.190 MVC was well tolerated in small animals, with no reported significant adverse events; it inhibited the cardiac potassium hERG channel only weakly and had no effect on the QTc interval.190,194 In cynomolgus monkeys, MVC did not affect various immune system parameters such as lymphocyte subsets, natural killer cell activity, phagocyte counts or oxidative activity; the IgM or IgG responses to keyhole limpet haemocyanin (KLH) were also unaltered, and MVC had no effect on the general histopathology of the immune system.195 In a review of six clinical studies involving 195 HIV-1-infected MVC recipients, the most commonly noted treatment-related adverse events were headache, dizziness, nausea, asthenia, flatulence, and rhinitis, mostly of mild or moderate severity, with incidence rates similar to those in placebo recipients.196 Four MVC recipients discontinued the study drug, three due to postural hypotension, the other because of elevated hepatic transaminases. There were no clinically significant changes in QTc interval in these six studies of HIV-1-infected recipients,196 or in a formal evaluation of single-dose MVC in 61 HIV-1-negative volunteers.197 A pharmacogenetic analysis revealed no association between 16 different CCR5 polymorphisms and blood pressure in 165 HIV-1-negative and 80 HIV-1-infected patients, suggesting that postural hypotension is not associated with CCR5 genotype.198 An intensive study in HIV-1-negative volunteers showed that MVC at a dose of 900 mg (three times the proposed 300 mg dose) had a mild vasodilatory effect by reducing systemic vascular resistance.199
A single case of severe hepatotoxicity leading to liver failure occurred in a study of MVC in treatment-naive patients.200 One subject randomized to receive MVC at 300 mg daily, together with zidovudine/lamivudine, developed a rash and severe hepatotoxicity after only four doses of the study drug. Although MVC was discontinued, isoniazid and other medications were continued and the patient was also started on lopinavir/ritonavir and parenteral paracetamol (acetaminophen). Hepatic transaminase levels worsened and the subject underwent liver transplantation on study day 16. An independent data safety monitoring board (DSMB) concluded that although MVC could not be excluded as the cause of hepatotoxicity, other medications are more likely to have been responsible. As a result, the MVC development program in both treatment-naive and treatment-experienced patients continues.200
Two randomized, placebo-controlled phase IIa studies were conducted to assess the short-term efficacy and safety of MVC in asymptomatic, HIV-1-infected patients with R5 viruses.173 In total, 82 individuals were randomized to receive MVC at 25, 100, or 300 mg once daily, 50, 100, 150 mg fed and fasted, or 300 mg twice daily, or placebo for 10 days. Their mean baseline plasma viral load was 4.62 log10 (42 000) copies/mL, with a mean CD4 cell count of 544/mm3. MVC resulted in a dose-dependent decrease in HIV-1 RNA levels at day 11, ranging from −0.43 log copies/mL (25 mg daily dose) to −1.60 log copies/mL (300 mg twice-daily dose) (Fig. 24-6). The mean CCR5 occupancy across the study group was >80% at day 5. The maximum viral load reduction occurred 1–4 days after MVC discontinuation. A single patient was retroactively identified as having entered the study despite the presence at baseline of R5+ X4 viruses (according to the Trofile assay); there was little change in HIV-1 RNA level during therapy. Viral tropism changes were observed in two other subjects, both receiving 100 mg of MVC daily. In both cases, R5+ X4 viruses were detected at day 11; in one case, only R5 viruses could be found at day 40; in the other, R5+ X4 viruses persisted through day 433. As noted above, the clinical and methodological significance of such observations remain to be determined. Although food reduced the MVC Cmax and AUC by 50–60%, Cmin was unchanged, and the viral load reductions were similar when 150 mg of MVC was given twice daily to fed or fasting volunteers. The investigators concluded that MVC was potent, generally well tolerated, and did not require food restrictions.173
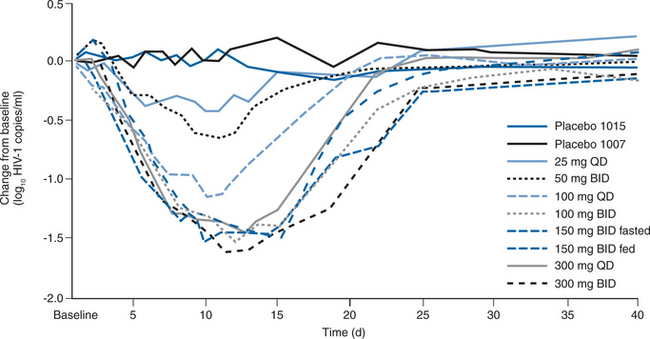
Figure 24-6 Mean change in log10 plasma viral load over time during treatment with maraviroc by dose group.
Reproduced from Fatkenheuer G, Pozniak AL, Johnson MA, et al. Efficacy of short-term monotherapy with maraviroc, a new CCR5 antagonist, in patients infected with HIV-1. Nat Med 11:1170, 2005.
Drug–drug interactions between MVC and other medications can be anticipated because it is a substrate of the hepatic cytochrome P450 system.201 A formal evaluation in HIV-1-infected patients taking various antiretroviral regimens and a single dose of 300 mg of MVC revealed that efavirenz-containing regimens reduced MVC concentrations by ∼50% (n = 16), lopinavir/ritonavir-containing regimens increased MVC concentrations by about twofold (n = 5) and nevirapine increased the MVC Cmax, but did not affect its AUC. In other studies, MVC concentrations were increased by CYP 3A4 inhibitors such as saquinavir, atazanavir, atazanavir/ritonavir, and ketoconazole, but MVC had no effect on the concentrations of zidovudine, lamivudine, tenofovir, trimethoprim, or oral contraceptive components.192 Decreasing MVC doses by half corrected the interactions with ritonavir or ritonavir-boosted PIs (except tipranavir). MVC concentrations were decreased by CYP 3A4 inducers such as efavirenz (40–50%) and rifampin (30%), but these reductions could be overcome by doubling the MVC dose. Using lopinavir (or saquinavir)/ritonavir and efavirenz with MVC approximately halves the pharmacokinetic effect of the inhibitors. A more recent study in 12 HIV-1-negative subjects showed there was no significant interaction between MVC and tipranavir/ritonavir, and that no dose adjustment was necessary.202
In two phase III studies of treatment-experienced subjects with R5 viruses, MVC, when added to an optimized background antiretroviral regimen, demonstrated significantly better virologic activity than placebo with 41–48% of subjects reducing HIV RNA to <50 copies/ml at 24 weeks in the MVC groups203,203a These results led to FDA approval of maraviroc for treatment-experienced patients in August 2007. In a phase II clinical study however, maraviroc showed no virologic activity in subjects with X4, R5X4 or indeterminate viral phenotype.203b A phase III study in treatment-naive subjects compared zidovudine/lamivudine combined with either MVC given twice daily or efavirenz and found that 65% (MVC) vs. 69% (EFV) had HIV RNA levels suppressed to <50 copies/ml at 48 weeks, a difference that could not exclude a pre-specified 10% difference required to show non-inferiority of MVC.203c

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


