Inherited Mixed Dyslipoproteinemias
Familial combined hyperlipidemia
The notion of familial combined hyperlipidemia (FCH, FCHL or multiple phenotype disease or multiple lipoprotein-type hyperlipidemia) evolved in the early 1970s from the work of Goldstein (who coined the term), Hazzard and Rose in the USA and from that of Nikkilä in Europe and their co-workers. Genetic analysis of lipid levels in families of survivors of myocardial infarction who had hyperlipidemia (1.40) revealed that, within a kindred, affected individuals (roughly 50% of the relatives) may have different lipoprotein phenotypes and that the phenotype may change over time within an individual. The lipoprotein phenotypes characterized according to the Fredrickson classification were either isolated hypercholesterolemia reflecting an increase in low-density lipoprotein-cholesterol (LDL-C) (type IIa), isolated hypertriglyceridemia due to increased very-lowdensity lipoproteins (VLDL) (type IV) or a combination of both (type IIb). Later, the presence of chylomicronemia was noted in some family members (type V) and it became clear that plasma apolipoprotein B (apoB) levels were elevated in this condition with a decrease in LDL particle size and an increase in LDL particle numbers (presence of atherogenic small dense LDL (sdLDL) prone to oxidation). This led Sniderman in Montreal to redefine FCHL as a hypertriglyceridemic hyper-apoB state. Hyper-apoB may be associated with normal cholesterol and triglyceride levels (referred to as ‘NB’ phenotype by the authors’ research group). Originally, the lipoprotein pattern was deemed inherited as an autosomal dominant trait (3.1) with reduced penetrance and/or delayed expression, but today a polygenic mode of inheritance is favoured. The etiology is not fully established as efforts have been baffled by several confounding elements such as soft diagnostic criteria, differences in proband ascertainment, phenotypic overlap (metabolic syndrome, familial dyslipidemic hypertension, atherogenic lipoprotein phenotypes), heterogeneity of disease, genetic variations across populations, gene and environment interactions and lack of a specific marker. Controversial issues in FCH have been critically reviewed by Aguilar Salinas and colleagues recently. FCH bears OMIM No. 44250 (www.ncbi.nlm.nih. gov/entrez/dispomim.cgi?id=144250).
FCH is a common form of primary hyperlipidemia. Its prevalence ranges between 1:50 and 1:30 in Caucasians and it accounts for 15-20% of patients with angiographically documented coronary artery disease (CAD) before 60 years of age. It may account for 10-15% of myocardial infarctions in Caucasians. Its atherogenic potential is based on the presence of a large number of LDL particles of smaller size that are more prone to oxidation and promote foam cell formation. Elevated plasma apoB alone allows the separation of a subset of CAD patients with ‘normal’ LDL-C from subjects with normal coronary arteries (3.2). A case-control comparison in the National Heart, Lung and Blood Institute (NHLBI) Family Heart Study found that both FCH and familial endogenous hypertriglyceridemia (FEHTG) were associated with an odds ratio for CAD of 2.0 (P = 0.003 and P = 0.002, respectively). Furthermore, both were associated with an increased prevalence of the metabolic syndrome of 65% and 77%, respectively, compared with control families (19%, odds ratio 3.3 [P < 0.001]).
Several metabolic abnormalities have been reported in FCH including hepatic apoB overproduction resulting in high plasma levels of apoB and small dense LDL, delayed postprandial chylomicron remnant clearance and prolonged postprandial elevation of plasma free fatty acids and apoB-48 (3.3). It has been inferred that the heterozygous state for lipoprotein lipase (LPL) deficiency could contribute to the FCH phenotype because reduced LPL activity has been observed in 30-50% of cases of FCH. A defective adipose tissue metabolism has also been postulated from several observations (3.4). These include a defect in the ability of insulin to suppress free fatty acid release from adipose tissue, impairment in insulin-mediated glucose disposal, an inefficient acylation stimulating protein (ASP)-mediated adipocyte triglyceride synthesis, and a defect in hormonesensitive lipase-mediated lipolysis. Lowered plasma levels of adiponectin have also been linked to the pathogenesis of FCH (3.4 and 3.5). The atherogenic potential is attributed to the increased number of small dense LDL particles that are prone to oxidation and favour foam cell formation. Plasma oxidized LDL are increased in FCH and there is a relative reduction in the cholesterol content of high-density lipoproteins (HDL). Triglyceride enrichment of HDL particles and enhanced hepatic lipase activity appear to be responsible for the reduction of HDL-C and HDL2-C in FCH. Subjects with predominantly sdLDL show a hypertriglyceridemic, low HDL-C phenotype, with moderately elevated apoB and LDL-C levels. This associates with a ten times higher number of VLDL1 particles as well as smaller VLDL2 particles, in combination with increased plasma insulin concentration compared with the hypercholesterolemic phenotype with more buoyant LDL particles. It was also shown recently that, independently of the lipoprotein phenotype, a general pattern of activated blood coagulation and impaired fibrinolysis (elevated thrombin-antithrombin complex [TAT], activated factor XII [F XIIa], von Willebrand factor [vWF], plasminogen activator inhibitor-1 [PAI-1] and tissue derived plasminogen activator [t-PA] values) is present in FCH. An increase in PAI-1 and soluble thrombomodulin is especially associated with features of the metabolic syndrome. A state of endothelial activation is also observed in FCH. Interestingly, mean carotid intima-media thickness (IMT) correlates positively with the vWF antigen in FCH.
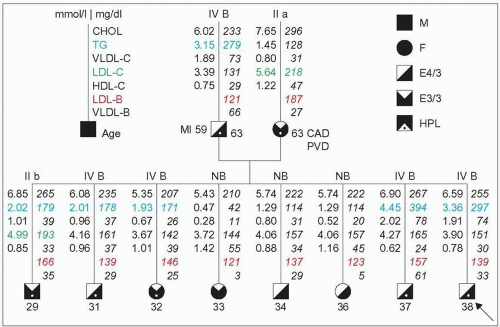 3.1 Apparent dominant mode of inheritance in a family with familial combined hyperlipidemia (FCH). This unique pedigree originates from a hyperlipidemic (HLP, marked by a dot on the diagram) father with the type IV phenotype and a hyperlipidemic mother with the type IIa phenotype (low-density lipoprotein cholesterol [LDL-C] in green >90th percentile). Both have elevated LDL–apoB (value of LDL-B in red) and both are presumed to originate from a familial combined hyperlipidemia family. At 63 years of age both have manifestations of atherosclerosis: myocardial infarction (MI), coronary artery disease (CAD) or peripheral vascular disease (PVD). The type IV is labelled B (IV B) because of the hyperapobetalipoproteinemia. This putative FCH × FCH mating is further borne out as all descendants of this couple have hyper-apoB defined by levels of total apoB ≥120 mg/dl (LDL-B + VLDL-B) or LDL–apoB ≥100 mg/dl. Multiple phenotypes are present in first-degree relatives. Five subjects have hypertriglyceridemia, defined by a fasting plasma value ≥150 mg/dl (1.7 mmol/l) (figures in blue). Three normolipidemic descendants have only hyperapobetalipoproteinemia (NB). This is compatible with a dominant mode of inheritance (as opposed to a co-dominant mode) because none of the eight offspring has a more severe lipoprotein phenotype than the others although the likelihood of including a homozygote is high (¼). The vertical transmission is obvious. Furthermore, the offspring range in age from 29 to 38 years of age, and there is a trend for the triglyceride levels to be higher in hypertriglyceridemics with ageing compatible with the reported delayed expression of this trait. The apoE phenotype is provided. It has been shown that the E4/3 phenotype (father and the four sons) may be associated with higher LDL-C levels than the E3/3 phenotype. |
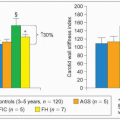
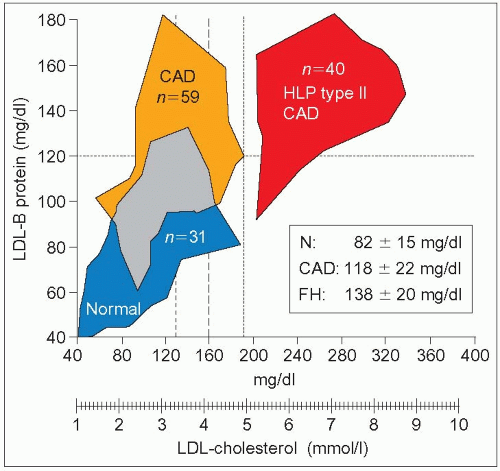 3.2 Hyper-apoB discriminates for coronary artery disease beyond low-density lipoprotein-cholesterol (LDL-C). This study was carried out in consecutive patients undergoing diagnostic cardiac catheterization for coronary artery disease (CAD). Thirty-one were ‘free of CAD’ (<25% stenosis) (blue cohort marked Normal) and 59 had CAD (≥50% stenosis; blinded evaluation, orange group). The mean ± SD for LDL-C was 2.9 ± 0.8 mmol/l (112 ± 30 mg/dl) and 3.46 ± 0.7 mmol/l (134 ± 27 mg/dl), respectively. A third group of 40 CAD subjects with ‘type II hyperlipoproteinemia’ representing essentially patients with familial hypercholesterolemia (FH) and an LDL-C ≥4.91 mmol/l (190 mg/dl) was used for comparison; the mean LDL-C in this group was 6.65 ±1.4 mmol/l (250 ± 55mg/dl, red). Although there is an overlap (grey zone) between normal and CAD subjects, discriminant analysis showed that LDL–apoB (LDL-B) most clearly separated the two groups compared with cholesterol, LDL-C or triglycerides. Although assessment of disease severity has the limitations of angiographic evaluation, this work supports the importance of plasma LDL–apoB to separate patients at risk for CAD in the presence of ‘normal’ LDL-C. Among the hyper-apoB patients most would fit the definition of familial combined hyperlipidemia (FCH). LDL-B levels in these patients reached values observed in familial hypercholesterolemia (FH) with much higher LDL-C concentrations. The upper limit of normal for LDL-C when this work was published was 4.91mmol/l (190 mg/dl). Afterwards it was brought down to 4.18mmol/l (160 mg/dl), and later to 3.36mmol/l (130mg/dl). Even with these new cut-offs, part of the CAD sample remains in the upper left quadrant of hyperapoB without hypercholesterolemia. Setting the limit of LDL-C in secondary prevention of CAD to 2.6 mmol/l (100 mg/dl) may include most of these hyper-apoB subjects and also most of the normal subjects. The apoB level is particularly important to know when identifying subjects at risk in an FCH family when triglycerides and total or LDL-C are normal (NB phenotype). The mean levels of LDL-B are given for the three different groups of patients illustrated on the left side of the graph. This figure was constructed from figures in the article by Sniderman AD et al. (1980). Proc Natl Acad Sci USA, 77: 604, linking the data point at the extremes of the distribution for each sample. |
A large effort has been made to identify the genetic defect responsible for FCH. Genome-wide linkage studies indicate that more than three genes are responsible for the lipoprotein pattern. Three chromosomal regions have been proposed for the location of these genes with strong supportive evidence:
1q21-23: an association was found with FCH in Finnish, German, US, Mexican and Chinese families; this region includes the APOAII gene but a close association was established with the upstream transcription factor 1 (USF1 gene) in the same region which regulates several genes of glucose and lipid metabolism; a single nucleotide polymorphism (SNP) in an intronic response element of this gene (usf1s2) appears to relate with the cardiovascular risk associated with FCH
11p14.1-q12.1: this region associates with the cholesterol and triglyceride traits. Recent combined linkage and association analyses point strongly to two separate alleles at the APOA1/C3/A4/A5 gene cluster on chromosome 11q23 to account for the transmission of FCH (Marc R et al. [2004] Circ Res, 94: 993-999)
16q22-24.1: this region associates with the low HDL-C trait. Other loci with weaker evidence include 1p31, 1q41, 6q16.1-16.3 and 8p23.3-22.
At present, the evidence supports the notion that FCH is an oligogenic disease with a complex pattern of inheritance involving a number of modifying genes.
The diagnosis of FCH is based on the demonstration in the patient’s family of the various lipoprotein phenotypes (IIa, IIb, IV, VB and NB, B indicating increased plasma apoB) which may change with time in an affected subject. Clinical features are usually discrete and non-specific such as xanthelasma and arcus corneae (3.6,3.7,3.8), until the manifestations of atherosclerosis, especially CAD, appear in the fifth and sixth decades in men, or the sixth and seventh decades in women. Cholesterol and/or triglycerides are elevated above the 90th percentile. Plasma apoB levels are usually ≥120 mg/dl in affected individuals. There is evidence that using apoB criteria for diagnosis may improve assignment to affected or non-affected. A nomogram based on values for total cholesterol, triglycerides and apoB adjusted for age and sex has been proposed by Veerkamp and co-workers for the diagnosis of FCH when at least one family member exhibits the phenotype and one family member has premature CAD. Until a clear genetic marker is available, however, a degree of uncertainty will remain in separating affected from nonaffected individuals.
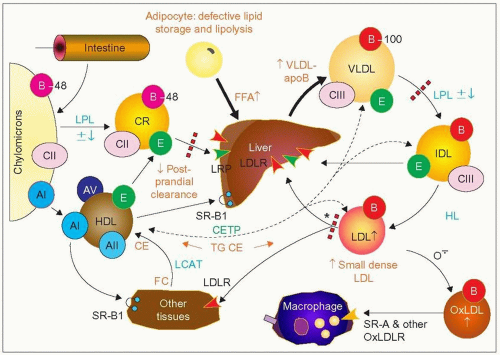 3.3 Metabolic defect in familial combined hyperlipidemia (FCH). In FCH there is a delayed clearance of postprandial chylomicron remnants (dotted red line on the left), with increased circulating levels of free fatty acids (FFA) and apoB-48. There is also reduced triglyceride synthesis from FFA in adipose tissue (see 3.4) resulting in an enhanced flux of FFA to the liver for triglyceride synthesis. The major metabolic defect in FCH is an overproduction of very-low-density lipoprotein (VLDL)-apoB by the liver leading to increased plasma VLDL and apoB concentrations. The VLDL particle size and lipid content are smaller than in familial endogenous hypertriglyceridemia (FEHTG). The increase in VLDL may account for the hypercholesterolemia as well as for the hypertriglyceridemia (IIb) but the final lipoprotein phenotype may depend on variations in the fate and or composition of the VLDL particles. Effective lipolysis coupled with reduced clearance of LDL may result in isolated hypercholesterolemia (IIa). In contrast, reduced lipoprotein lipase (LPL) activity (LPL ± ↓, dotted red line on the top right) — which occurs in as many as 50% of cases of FCH — with effective LDL clearance may favour the hypertriglyceridemic phenotype (IV), sometimes with traces of chylomicrons (type V). Phenotype variation over time may result from the transient effect of modulating environmental or genetic factors. The LDL particles formed tend to be small, dense and numerous. This is determined in part by the concentration and size of triglyceride-rich lipoproteins as there is an inverse relationship between triglycerides and LDL particle size (r = —0.71, P < 0.001, Vakkilainen J, Jauhiainen M, Ylitalo K, Nuotio IO, Viikari JSA, Ehnholm C, Taskinen MR [2002]. LDL particle size in familial combined hyperlipidemia: effects of serum lipids, lipoprotein-modifying enzymes, and lipid transfer proteins. J Lipid Res, 43: 598-603. Small dense LDL (sdLDL) particles are made with the concerted action of cholesteryl ester transfer protein (CETP) and hepatic lipase (HL). Although an impaired LDL removal may be a secondary component in FCH pathophysiology (* with dotted red line), one family has been reported in which the FCH clinical and biochemical phenotype was essentially associated with a defect at this step in the absence of VLDL–apoB overproduction. For more details regarding the metabolic scheme and abbreviations, see 1.2 and 2.12. |
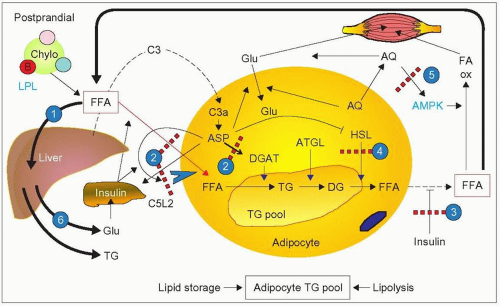 3.4 Putative involvement of adipose tissue in familial combined hyperlipidemia (FCH). In healthy subjects, there is a fine balance between free fatty acid (FFA) uptake and release by adipose tissue which reflects lipid storage and lipolysis, respectively, two physiologically important components of energy balance. In FCH, impairment of both lipid storage and lipolysis have been postulated. (1) After a fat meal in FCH, there is no major change in lipoprotein lipase (LPL) activity but increased serum free fatty acid (FFA) levels and flux to the liver with a transient increase in ketone bodies (0—8 hours). In the post-absorptive period (8—24 hours), FFA and ketone bodies rapidly decrease in subjects with FCH compared with normal subjects. This was ascribed by Meijssen et al. (2000), from Erasmus University, to inadequate incorporation of FFA into triglycerides in adipocytes coupled with a diminished release of FFA by hormone-sensitive lipase (HSP). This scenario is supported by other lines of evidence. (2) Acylation-stimulating protein (ASP), a complement C3a derivative, C3a-des-Arg, made in adipose tissue (with the concerted action of adipsin [factor D], complement factors C3 and B), promotes triglyceride (TG) synthesis by stimulating acyl-CoA:diacylglycerol acyl-transferase (DGAT), inhibits hormonesensitive lipase (HSL) and favours glucose uptake in adipocytes. ASP is a lipid-storage hormone. Some of its autocrine and paracrine effects are mediated by its receptor C5L2 (an orphan G protein-coupled receptor) and enhanced by insulin. ASP activity is impaired in FCH and ASP-mediated triglyceride synthesis is reduced in adipose cells from FCH subjects as shown by Cianflone and co-workers in Montreal. They have also shown that in fibroblasts from FCH patients (patients with ‘hyperapobetalipoproteinemia’) the specific binding of ASP to a single class of surface receptors was reduced in proportion to reduction in triglyceride synthesis (apart from evidence of reduced receptor number, it is not established whether there is impaired receptor-ligand interaction or ASP resistance in FCH). This results in a diversion of FFA from lipid storage to liver uptake for triglyceride and very-low-density lipoproteins (VLDL) synthesis. The major source of C3, the precursor of ASP, is the liver. An increase in circulating C3 has been reported in FCH. (3) There is also a defect in the ability of insulin to suppress free fatty acid release from adipose tissue in FCH. (4) This adds to the problem of impaired activity of HSL leading to impaired lipolysis as reported by Reynisdottir and co-workers, from Huddinge Sweden, in 1995 in work carried out with subcutaneous adipose tissue from FCH patients, (although, in contrast, there has been no demonstration of reduced HSL gene expression nor linkage with the HSL gene in affected Finnish subjects). An inhibition of ASP receptor by an expanding triglyceride pool was postulated. (5) Adiponectin (AQ) is another hormone formed in adipose tissue with protective effects against atherosclerotic cardiovascular disease. van der Vleuten and colleagues in Nijmegen, the Netherlands, found that adiponectin is decreased in FCH independent of waist circumference and insulin resistance. It is an independent predictor of the atherogenic lipoprotein profile of FCH, including high triglyceride levels, low HDL-C levels and amount of circulating small dense LDL. Several properties of adiponectin may account for this. Adiponectin enhances fatty acid oxidation (FA ox) in the circulation and in skeletal muscle, through activation of AMP-activated kinase (AMPK). There is also a strong relationship between adiponectin levels and LPL activity. This is consistent with the fact that plasma apoB concentrations are inversely related to adiponectin levels and that adiponectin correlates positively with apoB catabolism (but not with apoB production) (3.5). Furthermore, adiponectin increases glucose (Glu) uptake by skeletal muscle and enhances insulin sensitivity. Therefore a reduction in adiponectin would enhance insulin resistance, which may occur in FCH. (6) Finally, FFA may also promote insulin resistance by increasing gluconeogenesis in the liver. Increased glucose production stimulates the pancreas to secrete more insulin, which may lead to increased fasting insulin and insulin resistance coupled with defective insulin action (see point 3). This can lead to reduced FFA trapping and further increase in circulating FFA thereby creating a vicious cycle. |
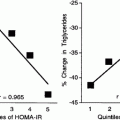
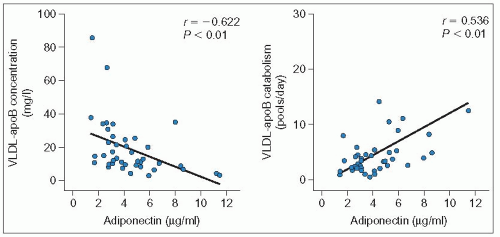 3.5 Relationship between adiponectin and very-low-density lipoprotein (VLDL)-apoB levels or catabolism. Ng and co-workers (2005) investigated the relationship of plasma adiponectin concentrations with VLDL-apolipoprotein B (apoB)-100 kinetics in men using stable isotopes. They showed that in multivariate analysis, plasma adiponectin concentration was the most significant predictor of plasma VLDL–apoB concentration (P = 0.001) and, with total or subcutaneous adipose tissue mass, was an independent predictor of VLDL–apoB catabolism (P < 0.001). The associations using a stepwise regression model including HOMA (homeostasis model assessment) score, age and total adipose tissue mass as co-variates are depicted in the left panel for apoB levels and the right panel for apoB catabolism (n = 41). There was no association of adiponectin with apoB production. In contrast with adiponectin, insulin resistance, measured with the HOMA, was a significant predictor of apoB production (P < 0.05). Interestingly, leptin, resistin, interleukin-6 and tumour necrosis factor a were not associated with any of the measured variables. Reproduced with permission from Ng TWK et al. (2005). Adipocytokines and VLDL metabolism: independent regulatory effects of adiponectin, insulin resistance, and fat compartments on VLDL-apolipoprotein B-100 kinetics? Diabetes, 54: 795-802. |
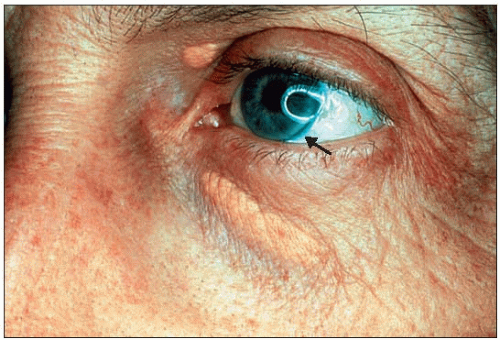 3.6 Discrete clinical signs in familial combined hyperlipidemia (FCH): xanthelasma and arcus corneae. Clinical signs when present tend to be discrete in FCH. Besides manifestations of atherosclerosis that might emerge eventually, such as arterial bruits, angina pectoris, transient ischemic attacks or other forms of expression of tissue ischemia, xanthelasma of the eyelids or arcus corneae may be the only clinical clue to the existence of FCH. This figure (the arrow is pointing to the arcus) is of a 46-year-old patient with hypercholesterolemia (phenotype IIa). |
Using current criteria, a diagnosis of FCH may not be made unless a family survey is carried out. Therefore, ‘combined hyperlipidemia’ or ‘mixed hyperlipidemia’, referring to the co-existence of hypercholesterolemia and hypertriglyceridemia in a patient, are not synonymous with ‘familial combined hyperlipidemia’. This combination could be due to a large number of causes including familial hypercholesterolemia (see 2.16) or polygenic hypercholesterolemia with inheritance of familial hypertriglyceridemia from one of the parents, dysbetalipoproteinemia type III, secondary causes of hypertriglyceridemia or hypercholesterolemia associated with or superimposed on an inherited form of dyslipidemia, etc. Tendon xanthomas are not present in FCH, unless FH co-segregates in the same family. As discussed in Chapter 2, FEHTG differs from FCH in that the VLDL of FEHTG are more enriched in lipids and of larger size than VLDL of FCH (see 2.15). Several risk factors seem to associate readily with FCH, such as obesity, hypertension and insulin resistance. Some degree of overlap may be seen with other conditions including the metabolic syndrome, familial dyslipidemic hypertension (FDH) and the atherogenic lipoprotein pattern B. It is not clear at present if FDH constitutes a separate clinical entity from FCH. As LPL mutations may segregate in FCH families especially in areas where a founder effect may have occurred, a mixed hypertriglyceridemia (type V) may be seen in patients homozygous for LPL deficiency as observed on occasion at the authors’ lipid clinic (3.9). This occurrence probably explains early reports of type V as one of the multiple phenotypes of FCH (3.10).
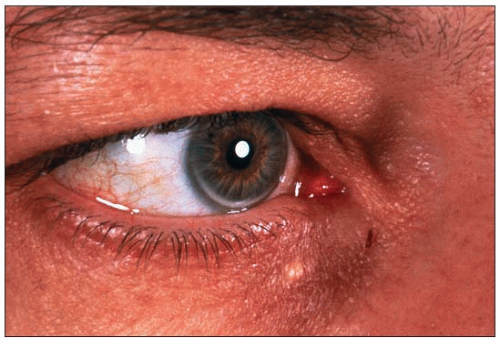 3.7 Discrete manifestations of familial combined hyperlipidemia (FCH): arcus corneae and xanthelasma. Corneal arcus and xanthelasma are not always as obvious as in 3.6. Instead of fully encircling the cornea (see 1.3), FCH is often manifested by a discrete lower (as above) or upper corneal crescent, always leaving a rim of clear cornea between the lipid deposit and the sclera. It may be easily missed if it is not looked for in particular using a light, especially for upper crescents, which are masked readily by the upper eyelid. They tend to occur more frequently in the elderly. In the past a distinction was made between arcus juvenilis and arcus senilis, the latter considered a degenerative ocular manifestation. In black people, arcus corneae are routinely seen in the absence of dyslipidemia. In this figure, a unique small xanthelasma is also present. Half of the subjects with xanthelasma also have dyslipidemia. Presence of either of these is an excellent clue for dyslipidemia. |
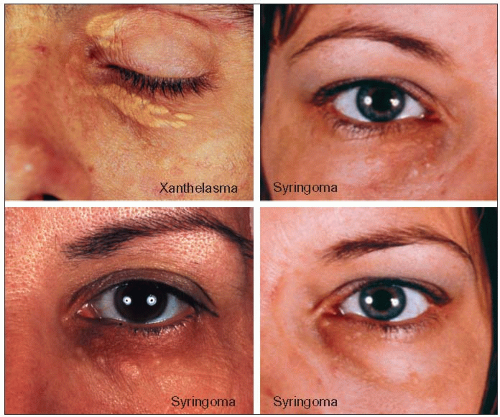 3.8 Syringoma is often mistaken for xanthelasma associated with dyslipidemia. The top left panel shows typical xanthelasmas of the eyelids and of the loose skin under the lower eyelid in a 38-year-old patient with familial combined hyperlipidemia (FCH; type IIa phenotype). They are typically planar, slightly raised, occurring in patches of various sizes, coalescing with a yellow to yellow-orange discoloration. The other three panels represent syringomas which, in contrast, are puncta of smaller sizes, are not found in patches or coalescing, and are more pinkish than yellow. These are small tumours, usually benign, of the sweat glands. The lower left panel shows typical syringomas in a 51-year-old woman, referred for hypercholesterolemia and xanthelasma of the eyelids. In fact she had histologically confirmed syringomas of the eyelids and not xanthelasma and hyperalphalipoproteinemia. Twenty-two years later her lipoprotein profile had changed very little: total cholesterol 5.98 mmol/l (231 mg/dl), low-density lipoprotein-cholesterol (LDL-C) 3.41 mmol/l (132 mg/dl), triglycerides 0.93 mmol/l (82 mg/dl), and HDL-C 2.15 mmol/l (83 mg/dl). She never developed clinical manifestations of atherosclerosis. The two panels on the right show a 33-year-old women who was referred to the authors’ lipid clinic in 1972 for ‘xanthelasma’ and hyperlipidemia discovered fortuitously during a routine clinical examination. Further evaluation revealed that she had syringoma of the eyelids and not xanthelasma. The modest rise in cholesterol levels to 6.85 mmol/l (265 mg/dl) and triglycerides to 2.74 mmol/l (243 mg/dl) was not due to a familial diathesis but was secondary to oral contraceptives (norethindrone 1 mg/mestranol 50 μg). Lipids normalized on withdrawal and the dyslipidemia recurred on rechallenge. The positive family history of a premature death at the age of 45 of a maternal aunt due to coronary causes, which had suggested a familial disease turned out to be that of an unrelated spouse of a family member. Correct interpretation of the clues suggestive of dyslipidemia is a key element of the diagnostic process. |
There is evidence that FCH may be heterogeneous. Aguilar Salinas and co-workers in 1997 reported a family in which the clinical phenotype is indistinguishable from that of FCH but instead of observing an overproduction of VLDL apoB, the metabolic defect was an impaired apoB catabolism (3.3).
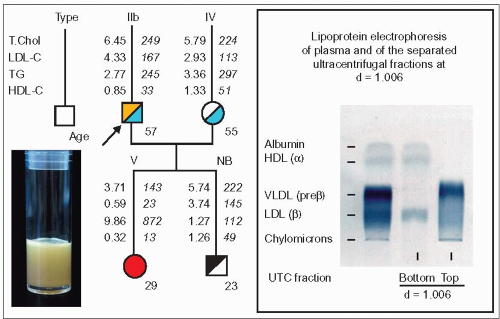 3.9 Type V secondary to the P207L LPL mutation in a familial combined hyperlipidemia (FCH) family. In this family the proband is a 57-year-old man with FCH. On the paternal side, four of his relatives had had a myocardial infarction and/or a cerebrovascular accident between the ages of 45 and 79. He himself had a cerebral thrombosis following a left carotid endarterectomy at the age of 44 after a few episodes of amaurosis fugax. His 55-year-old spouse was found to have hypertriglyceridemia (type IV). His daughter first consulted at the age of 21 years, five years after her father’s first visit to the authors’ lipid clinic. A diagnosis of familial hyperchylomicronemia had been made at the age of 8 months. She was followed regularly thereafter and was on a very low fat/no alcohol diet. Thus, she was able to avoid having pancreatic complications or developing eruptive xanthomas. Homozygosity for the P207L mutation of the LPL gene was established at the age of 19. At the authors’ lipid clinic her lipoprotein phenotype was found to be type V. Chylomicrons ranged from traces on lipoprotein electrophoresis to a typical creamy layer on top of a diffusely milky plasma (image on the left). Her triglycerides tended to increase over seven years of follow-up and fluctuated widely from 3.83 mmol/l to 21.7 mmol/l. The recent lipoprotein analysis given above shows the phenotypic profile of type V in which the very-low-density lipoproteins (VLDL) fraction predominates (right panel). Her 23-year-old brother is normolipidemic, but his total plasma apoB is 121 mg/dl, which is compatible with the presence of an FCH susceptibility gene at this age (type NB). Red, mixed hypertriglyceridemia; yellow, hypercholesterolemia; blue, hypertriglyceridemia; black, hyperapobetalipoproteinemia. |
The treatment of FCH is a function of the lipid abnormality using dietary measures that would be appropriate for hypertriglyceridemia, hypercholesterolemia or both and for associated cardiovascular risk factors. The same attitude applies for drug treatment when goals of therapy are not attained, which is often the case, with lifestyle changes alone: statins for hypercholesterolemia and fibrates for hypertriglyceridemia. The combined phenotype may need a combination of both drugs applied with caution and monitoring for potential side effects. Nicotinic acid and its sustained release forms may be effective in monotherapy if well tolerated.
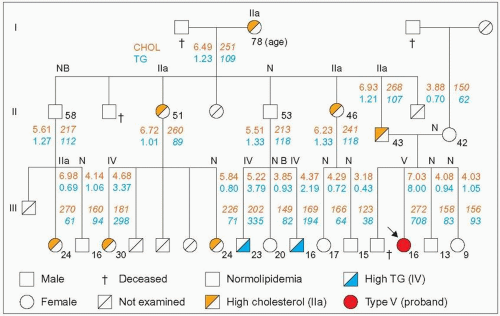 3.10 Mixed hypertriglyceridemia (type V) in a familial combined hyperlipidemia (FCH) family. There have been several reports of mixed hypertriglyceridemia (lipoprotein phenotype V) occurring in FCH families. This figure concerns one of the early reports (Kobori et al. 1986). The proband (arrow) was a 16-year-old girl with recurrent episodes of abdominal pain. As is often the case, laparotomy was carried out, which revealed that the pain was secondary to pancreatitis. Later on, the plasma was noted to be milky, which revealed the cause and she was referred to a lipid clinic where the family survey was done. This case is interesting because, first, the diagnosis of FCH was supported by raised levels of apoB in all hyperlipidemic cases, including the proband and the other hypertriglyceridemic subjects; there were two different phenotypes in the family (IIa and IV); there were discrete clinical manifestations (19 relatives were examined; there were no tendon xanthomas but corneal arcus was present in the father) and the presence of two normolipidemic subjects with hyper-apoB (NB phenotype). There was no overt coronary artery disease in this family with relatively young members (the oldest in generation II was 58 years and in generation III was 30 years). Second, there was a reduction in post-heparin lipolytic activities in the proband: LPL of 0.8 μM FFA/h/ml (normal: 2.5 ± 0.9) and HL 3.2 μmM FFA/h/ml (normal: 6.9 ± 3.1), which is compatible with the authors’ experience and that of others that mutations of the LPL gene may segregate independently in some FCH families. Secondary causes of dyslipidemia were excluded and there was no apoCII deficiency. The proband had the common E3/3 apoE phenotype. Her lipoprotein profile indicated endogenous hypertriglyceridemia (type IV) when her plasma triglycerides were lowered. There were no cases of dysbetalipoproteinemia (type III) in the family. Total cholesterol (dark orange figures, orange-yellow shading) and triglycerides (blue figures, blue shading) are given in mmol/l and in mg/dl. They are given in mg/dl in the original article. The lipoprotein phenotype is given at the top of the line of each pedigree member; note that ‘NB’ phenotype is the authors’ interpretation, the plasma apoB was 116 mg/dl for the 58-year-old and 90 mg/dl for the 20-year-old relative. The age is given in the lower right corner of the square for males and the circle for females. Redrawn and modified from Kobori S et al. (1986). A kindred of familial combined hyperlipidemia (FCHL) with proband showing type V hyperlipoproteinemia. Jpn J Med, 25: 306-312. |
Familial dysbetalipoproteinemia and remnant excess
Classical dysbetalipoproteinemia type III
Although uncommon, the importance of this disease stems from the following points:
it is a major diagnostic challenge (it is often missed)
it has distinct clinical features, some of them pathognomonic, which when present allow the clinician to make a conclusive diagnosis at bedside
complex interactions determine its expression
failure to make a diagnosis exposes the subject to premature CAD
treatment is effective and avoids catastrophic consequences.
 3.11 Three pioneers in lipidology. From left to right: Donald S Fredrickson, who developed the classification of hyperlipoproteinemia based on the lipoprotein phenotype; Gerd Assmann, who conducted the PROCAM epidemiological study; and Gerd Utermann, head of one of the two groups that unraveled the apoE polymorphism. This photograph was taken in October 1994 at the Xth International Symposium of Atherosclerosis in Montreal. |
Familial dysbetalipoproteinemia (type III hyperlipoproteinemia, type III, broad beta disease, remnant removal disease, floating β lipoprotein disease) is a relatively rare condition (frequency of about 1—5/5000; 1—5/10 000 in Japan). It was first recognized in the early days of lipoprotein separation by analytical ultracentrifugation by Gofman and co-workers in 1952, at the Donner Laboratory in California, as a distinct lipoprotein profile (increase in the Sf 12-20 and Sf 20-100 fractions) associated with tuberous xanthomas. However, it was not until 1967 that it was identified as a separate nosological entity by Fredrickson (3.11) and his group at the National Institutes of Health (NIH). Many tools were available at the time to study plasma lipoprotein metabolism (3.12). The diagnosis then was essentially based on the clinical findings coupled with the appearance of a ‘broad beta band’ on electrophoresis of plasma lipoproteins migrating where β-lipoproteins (LDL) migrate (3.13 and 3.14), and reflecting an increase in cholesteryl ester enriched particles. These are partially degraded normal VLDL and chylomicrons, referred to as remnants (or intermediate-density lipoproteins [IDL]), forming large buoyant particles, depleted in apoC and enriched in apoE. Although they float with the VLDL fraction when separated by ultracentrifugation, they migrate at the level of β-lipoproteins on lipoprotein electrophoresis (hence the term ‘floating β-lipoprotein’ or β-VLDL) (3.13). The accumulation of plasma remnants results in high levels of cholesterol, triglycerides and apoE, low LDL-C concentrations and normal or low HDL-C. Because remnants are enriched in cholesteryl esters, the VLDL-C/triglyceride ratio is typically high (>0.3, if mg/dl is the unit used, or >0.7, if mmol/l is the unit used). This abnormal ratio was a major diagnostic clue in the past, before the role of apoE polymorphism was uncovered in the 1970s in the laboratories of Breslow and Zannis in the USA and Utermann (3.11) in Germany, and the association with the apoE2/2 phenotype established in Utermann’s laboratory (1977). Thereafter, demonstration that the affected subject carries the E2/2 phenotype (3.15) or genotype (3.16) became a prerequisite to establish the diagnosis of type III.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


