Chapter Outline
INHERITED BONE MARROW FAILURE SYNDROMES ASSOCIATED WITH PANCYTOPENIA
INHERITED BONE MARROW FAILURE SYNDROMES ASSOCIATED WITH ISOLATED CYTOPENIAS
Inherited bone marrow failure syndromes (IBMFSs) are a diverse collection of genetic disorders associated with inadequate blood cell production. Bone marrow failure (BMF) may manifest as an isolated cytopenia (pure red cell aplasia, neutropenia, or thrombocytopenia) or as pancytopenia with the clinical picture of aplastic anemia. Table 7-1 summarizes the more common IBMFSs, the blood cell lineages affected, the genes mutated and their modes of inheritance, and the pathways involved. Other organ systems are often affected by these genetic abnormalities, resulting in birth defects or clinical disease in nonhematopoietic organs. Birth defects and extra hematopoietic manifestations are often characteristic and may be noticed before the onset of BMF or may be the only manifestations of disease. BMF may be present at birth (congenital BMF) or develop later in life. IBMFSs are not always inherited; the genetic mutations responsible may occur de novo during early embryonic development. Several, but not all, IBMFSs are characterized by a predisposition for malignant transformation, in particular of the hematopoietic system, including the development of myelodysplastic syndrome (MDS) and acute leukemia, but also for a variety of other forms of cancer (see Table 7-1 ).
| Syndrome | Inheritance Pattern | Peripheral Blood Manifestations | Associated Malignant Diseases | Gene | Chromosomal Location | Affected Pathway |
|---|---|---|---|---|---|---|
| Fanconi anemia (FA, MIM #227650) | AR | Pancytopenia | MDS/AML Squamous cell carcinoma Other tumors | FANCA | 16q24.3 | DNA repair, homologous recombination |
| XLR | FANCB | Xp22.31 | ||||
| AR | FANCC | 9q22.3 | ||||
| AR | FANCD1/BRCA2 | 13q12-13 | ||||
| AR | AML Wilms tumor Medulloblastoma | FANCD2 | 3p25.3 | |||
| AR | MDS/AML Squamous cell carcinoma Other tumors | FANCE | 6p21.3 | |||
| AR | FANCF | 11p15 | ||||
| AR | FANCG/ XRCC | 9p13 | ||||
| AR | FANCI | 15q26.1 | ||||
| AR | FANCJ/BACH1/ BRIP1 | 17q22-q24 | ||||
| AR | FANCL | 2p16.1 | ||||
| AR | FANCM | 14q21.3 | ||||
| AR | Wilms tumor Medulloblastoma | FANCN/ PALB2 | 16p12.2 | |||
| AR | ? | FANCO/ RAD51C | 17q22 | |||
| AR | ? | FANCP/ SLX4 | 16p13.3 | |||
| Dyskeratosis congenita (DC) | XLR (MIM, #305000) | Pancytopenia | MDS/AML Squamous cell carcinoma Other tumors | DKC1 | Xq28 | Telomere maintenance, ribosome biogenesis |
| AD (MIM #127550) | TERC | 3q26.2 | Telomere maintenance | |||
| AD (MIM #613989) | TERT | 5p15.33 | Telomere maintenance | |||
| AD (MIM #613990) | Unknown | TINF2 | 14q11.2 | Telomere maintenance | ||
| AR (MIM #224230) | Unknown | NOP10/ NOLA3 | 15q14-q15 | Telomere maintenance, ribosome biogenesis | ||
| AR (MIM # 613987) | Unknown | NHP2/ NOLA2 | 5q35.5 | Telomere maintenance, ribosome biogenesis | ||
| AR (MIM # 613988) | Unknown | WDR79/TCAB1/WRAP53 | 17p13.1 | Telomere maintenance | ||
| AR (MIM # 612199) | Unknown | CTC1 | 17p13.1 | Telomere maintenance | ||
| AR (MIM #615190) | Unknown | RTEL1 | 20q13.3 | DNA repair, homologous recombination, telomere maintenance | ||
| Shwachman-Diamond syndrome (SDS, MIM #260400) | AR | Neutropenia with progression to pancytopenia | MDS/AML | SBDS | 7q11.21 | Ribosome biogenesis, ribosome assembly |
| Cartilage-hair hypoplasia (CHH, MIM #250250) | AR | Neutropenia Lymphopenia Anemia | Lymphoma Basal cell carcinoma | RMRP | 9p13.3 | Mitochondrial DNA replication, ribosome biogenesis, degradation of CLB2 mRNA |
| Pearson marrow-pancreas syndrome (PS, MIM #557000) | Mitochondrial | Neutropenia with progression to pancytopenia | None | mt DNA deletion | Mitochondrial function | |
| Diamond-Blackfan anemia (DBA, MIM #105650) | AR | Anemia with rare progression to pancytopenia | MDS/AML * Osteosarcoma | RPS19 | 19q13.2 | Ribosome biogenesis |
| RPS24 | 10q22-23 | |||||
| RPS17 | 15q25.2 | |||||
| RPS10 | 6p21.31 | |||||
| RPS26 | 12q13.2 | |||||
| RPS7 | 2p25.3 | |||||
| RPS29 | 14q21.3 | |||||
| RPL35a | 3q29-qter | |||||
| RPL5 | 1p22.1 | |||||
| PRL11 | 1p36.1-35 | |||||
| RPL26 | 17p13.1 | |||||
| RPL15 | 3p24.2 | |||||
| XLR | Anemia and neutropenia | GATA1 | Xp11.23 | |||
| Congenital dyserythropoietic anemia (CDA) type I (MIM #224120) | AR | Anemia | None | CDAN1 † | 15q15.1-15.3 | Unknown |
| CDA type II (MIM #224100) | AR | None | SEC23B (CDAN2) | 20q11.23 | Unfolded protein response | |
| CDA type III (MIM #105600) | AD | Myeloma | KIF23MKLP1 | 15q21-25 | Cytokinesis | |
| CDA type IV (MIM #613673) | AD | KLF1 (CDAN4) | 19p13.2 | Erythroid transcription factor | ||
| Severe congenital neutropenia due to ELANE mutation (SCN, MIM #202700) | AD | Neutropenia | MDS/AML | ELANE | 19p13.3 | Unfolded protein response (?) |
| Cyclic neutropenia (CN, MIM #162800) | AD | Neutropenia | None | ELANE | 19p13.3 | Unfolded protein response (?) |
| Kostmann syndrome (MIM #610738) | AR | Neutropenia | MDS/AML | HAX1 | 1q21.3 | Regulation of apoptosis (?) |
| Severe congenital neutropenia due to G6PC3 mutation (MIM #612541) | AD | Neutropenia | MDS/AML | G6PC3 | 17q21.31 | Glucose metabolism, glycosylation (?), unfolded protein response |
| Severe congenital neutropenia due to GIF1 mutation (MIM #613107) | AD | Neutropenia Lymphopenia | MDS/AML | GIF1 | 1p22.1 | Transcription repressor of ELANE |
| Severe congenital neutropenia due to CSF3R mutation | AD Sporadic | Neutropenia | MDS/AML | CSF3R | 1p34.3 | GCSF receptor signaling |
| Barth syndrome (BTHS, MIM #302060) | AR | Neutropenia | None reported | TAZ1 | Xq28 | Changes in mitochondrial architecture and function |
| Cohen syndrome (COH1, MIM #216550) | AR | Neutropenia | None reported | COH1 | 8q22-q23 | Lysosomal protein trafficking |
| Chédiak-Higashi syndrome (CHS1, MIM #214500) | AR | Neutropenia | None reported | CHS1/LYST | 1q42.3 | Lysosomal protein trafficking |
| Griselli syndrome (GS2, MIM # 607624) | AR | Neutropenia Thrombocytoepnia | None reported | RAB27A | 15q21.3 | Lysosomal function (?) |
| Hermansky-Pudlak syndrome 2 (HPS2, MIM #608233) | AR | Neutropenia | None reported | AP3B1 | 5q14.1 | Lysosomal protein trafficking, unfolded protein response (?) |
| Immunodeficiency due to defect in MAPBP-interacting protein P14 (MIM #610798) | AR | Neutropenia | None reported | MAPBPIP | 1q22 | Lysosome biogenesis |
| Glycogen storage disease 1b (GSD1B, MIM #23220) | AR | Neutropenia | Renal carcinoma | G6PT1 | 11q23.3 | Glycogen storage |
| Hyper-IgM immunodeficiency syndrome (XHIM, MIM #308230) | XLR | Neutropenia Pancytopenia | None reported | CD40LG (HIGM1) | Xq26.3 | B-cell activation |
| WHIM syndrome (MIM #19370) | AD | Neutropenia Lymphopenia | None reported | CXCR4 † | 2q22.1 | Neutrophil mobilization |
| Poikiloderma with neutropenia (MIM #604173) | AD | Neutropenia | MDS/AML Skin cancer | USB1 | 16q21 | Processing of U6 spliceosomal RNA |
| Congenital amegakaryocytic thrombocytopenia (CAMT, MIM #60448) | AR | Thrombocytopenia with progression to pancytopenia | AML | c-MPL | 1q35 | Megakaryopoiesis |
| Thrombocytopenia with absent radii (TAR, MIM # 274000) | AR | Thrombocytopenia | AML | RBM8A | 1q21.1 | Part of exon junction complex |
| Amegakaryocytic thrombocytopenia with radioulnar synostosis (ARTUS, MIM #605432) | AD | Thrombocytopenia with progression to pancytopenia | AML | HOXA11 | 7p15-p14.2 | Morphogenesis |
| Familial platelet disorder with associated myeloid malignancy (FPD/AML, MIM #601399) | AD | Thrombocytopenia | MDS/AML | AML1 | 21q22.3 | Hematopoietic cell differentiation |
| Wiskott-Aldrich syndrome (WAS, MIM #301000) | XLR | Thrombocytopenia | Lymphoma | WAS † | Xp11.23 | Loss of function, signal transduction from cell surface to actin cytoskeleton |
| X-linked severe congenital neutropenia (SCNX, MIM #300299) | Neutropenia Monocytopenia | MDS/AML | WAS † | Gain of function; disrupt autoinhibiton | ||
| X-linked thrombocytopenia (XLT, MIM #313900) | Thrombocytopenia | Lymphoma | WAS | Loss of function, signal transduction from cell surface to actin cytoskeleton | ||
| Congenital dyserythropoiesis associated with thrombocytopenia (MIM #300367) | XLR | Thrombocytopenia | None | GATA1 | Xp11.23 | Erythroid and megakaryocytic development |
| Rare Forms of Inherited Bone Marrow Failure Syndromes | ||||||
| Nijmegen breakage syndrome (NBS, MIM #251260) | AR | Pancytopenia | AML Lymphoma | NSB1 † | 8q21 | DNA repair |
| DNA ligase IV syndrome (LIG4, MIM #606593) | AR | Pancytopenia | Leukemia | LIG4 | 13q22-q34 | DNA repair |
| Seckel syndrome (SCKL1, MIM #210600) | AR | Pancytopenia (not genetically subtyped) | Leukemia Lymphoma Oropharyngeal cancer (not genetically subtyped) | ATR SKC1 | 3q22-q24 | DNA repair |
| PCNT SCKL4 | 21q22.3 | Centrosomal function, ATR signaling pathway | ||||
| AR | CENPJ SCKL4 | 13q12.12 | Centrosomal function, ATR signaling pathway | |||
| AR | CEP152 SCKL5 | 15q21.1 | Centrosomal function, ATR signaling pathway | |||
| AR | CtIP/RBBP8 SCKL2 | 18q11.2 | Centrosomal function, ATR signaling pathway | |||
| AR | ATRIP | 3p21.31 | Interacts with ATR, ATR signaling pathway | |||
| Dubowitz syndrome (MIM %223370) | AR | Pancytopenia | Leukemia Lymphoma Neuroblastoma Cancer | Unknown | Unknown | DNA repair (?) |
| Schimke syndrome (SIOD, MIM #242900) | AR | Pancytopenia | Lymphoma | SMARCAL1 | 2q34-q36 | Chromatin remodeling |
| Duncan/Purtilo syndrome (XPL, MIM #308240) | XLR | Pancytopenia | EBV lymphoma | SH2D1A/SAP | Xq25 | B-cell and T-cell function |
* No specfic ribosomal gene mutation has been assocated with AML/MDS.
The prevalence of IBMFSs is difficult to ascertain because BMF registries are biased by referral, founder effect, and the enrollment of mainly pediatric patients. The incidence of IBMFSs has been estimated to be 40 to 65 per 10 6 live births. Originally, IBMFSs were considered to be mainly diseases of childhood. In children 25% to 30% of BMF cases were thought to have a genetic cause, whereas among adults an inherited genetic cause was considered in only a minority of patients. However, more recent data suggest that the prevalence of IBMFS patients has probably been underestimated, particularly in adults. In addition, improved medical care after bone marrow transplantation (BMT) has extended life expectancy and shifted the management of IBMFS patients to adult hematologists.
Timely and correct diagnosis of an IBMFS is paramount because management of an IBMFS differs from that of acquired BMF. Treatment of IBMFSs often requires a complex interdisciplinary approach that includes hematologists, geneticists, hematopathologists, BMT specialists, oncologists, endocrinologists, orthopedists, neurologists, gastroenterologists, nephrologists, dentists, and other specialists. Differentiation between acquired and inherited BMF is of importance not only for affected individuals but also for their family members. Early and correct diagnosis of an IBMFS allows intensified and focused disease surveillance, anticipatory guidance, and avoidance of treatment-related toxicities. Identification of a gene mutation known to be associated with BMF can definitively establish the diagnosis before clinical symptoms advance and help identify an unaffected bone marrow donor in the family. It is therefore imperative that at the time of initial evaluation maximal effort be made to distinguish acquired from inherited forms of BMF.
Considerable advances have been made in the last 15 years in elucidating the genetic causes of IBMFSs (see Table 7-1 ). In the past investigations of IBMFSs were largely restricted to careful observations and descriptions of clinical manifestations in the patient and family, as well as meticulous study of the clinical course of the disease. However, innovations in genomic sequencing technology have led to the identification of a rapidly increasing number of genes that when mutated are responsible for certain IBMFSs. Characterization of these genes and investigation of their role in hematopoiesis are powerful new tools to explore the pathways responsible for BMF.
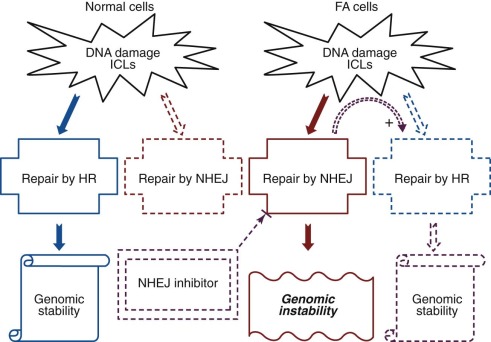
Traditionally, IBMFSs have been classified according to their clinical features. Increasingly this classification scheme is being supplanted by one based on the affected biochemical pathway (see Table 7-1 ). Surprisingly, the affected pathways often entail “housekeeping” functions important for most cell types (DNA repair, telomere maintenance, or ribosome biosynthesis) rather than functions unique to hematopoietic cells. Why hematopoiesis is preferentially and sometimes even exclusively affected by a derangement in these pathways is intriguing and the focus of current investigations in the field of BMF research. One feature these pathways share is that their derangement usually causes the activation of p53, suggesting that this molecule plays a central role in the pathogenesis of IBMFSs ( Fig. 7-1 ).
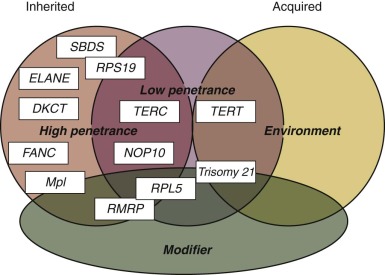
With the increased knowledge of how the genetic framework influences the development of BMF, distinguishing between inherited and acquired BMF has become more difficult. Previously, IBMFSs were restricted mainly to patients with BMF caused by gene mutations with high disease penetrance. Today, it is increasingly being recognized that, in addition, a growing number of patients have BMF caused by gene mutations that have low or very low penetrance, with disease developing only occasionally or just under certain precipitating circumstances. Thus the majority of patients in whom acquired idiopathic aplastic anemia was previously diagnosed may have an underlying genetic component. Figure 7-1 schematically illustrates the possible interrelationship between genetic mutation, environment, and gene modifiers in the pathogenesis of BMF. In the coming years, advances in DNA sequence technology are likely to bring a wealth of new information to the field of BMF research that in combination with the power of bioinformatics is likely to shift some of the current disease paradigms. The increased understanding of the genetic components and their role and importance in the pathogenesis of disease will allow us to improve the diagnosis, management, and treatment of patients with BMF.
Inherited Bone Marrow Failure Syndromes Associated with Pancytopenia
Fanconi Anemia
Fanconi anemia (FA) is a genetically and phenotypically heterogeneous disorder. The disease is named after the Swiss pediatrician Guido Fanconi (1892-1979), who first described the syndrome in 1927. Clinically, FA is characterized by a variety of congenital abnormalities, progressive BMF, and a propensity for the development of leukemia and other forms of cancer. Cells from FA patients have a striking hypersensitivity to DNA interstrand cross-links ; this trait has become the basis of a clinical diagnostic test for FA ( Box 7-1 ).
Fanconi anemia (FA, MIM #227650) is an autosomal recessive and X-linked disorder characterized by progressive bone marrow failure, congenital abnormalities, and a predisposition for malignant disease. Cells from patients with FA exhibit spontaneous chromosomal instability and a characteristic hypersensitivity to DNA interstrand cross-linking agents such as diepoxybutane (DEB) and mitomycin C (MMC).
Epidemiology
The incidence of FA is difficult to ascertain. The Fanconi Anemia Research Fund, Inc., estimates that about 30 persons with FA are born in the United States each year; this would indicate that the prevalence of FA is about 2.5 per million and the carrier frequency is approximately 1 in 180. The male-to-female ratio is 1.2 : 1. FA has been reported in all races and ethnic groups. The world’s highest prevalence of FA, caused by a founder effect, is seen in Spanish gypsies, who have a carrier frequency of 1 in 70. A high prevalence of FA is also found in individuals of Ashkenazi Jewish decent, who have an approximate heterozygous frequency of 1 in 77, and in South African Afrikaans, in whom carrier frequency is estimated to be 1 in 83.
Clinical Manifestations
The clinical manifestations of FA are heterogeneous (variable penetrance and expressivity; for definitions, see Box 7-2 ). Affected family members can have a wide variety of abnormalities (variable expressivity). Even monozygotic twins have been described to differ in their physical and hematologic manifestations. However, the occurrence of malformations is nonrandom. Siblings are usually concordant for the presence or absence of multiple congenital abnormalities; likewise, the age at onset of hematologic manifestations is similar within an individual family. Thus the variability in clinical findings appears to be to some extent linked to the different genotypes. The emergence of revertant cells leading to hematopoietic mosaicism further contributes to disease variability (see the section on Somatic Mosaicism, Functional Reversion, and Attenuation of Fanconi Anemia Cells). Originally, the diagnosis of FA was based on the presence of both aplastic anemia and a specific, though broad, range of physical abnormalities. With the availability of more sensitive and specific tests for the diagnosis of FA, including the availability of genetic testing, it has become evident that a significant proportion of patients lack congenital abnormalities (25% to 40%) or fail to develop aplastic anemia. Nevertheless, these individuals are still at risk for late complications such as leukemia and cancer.
Heritability, penetrance, and expressivity are distinct but related concepts. Heritability is the number of family members who have inherited a genotype versus the number of family members who have not. Penetrance is defined by the number of individuals who show a phenotype versus the number of individuals who carry the genotype. Expressivity measures the variability and severity of an expressed phenotype.
Congenital Abnormalities
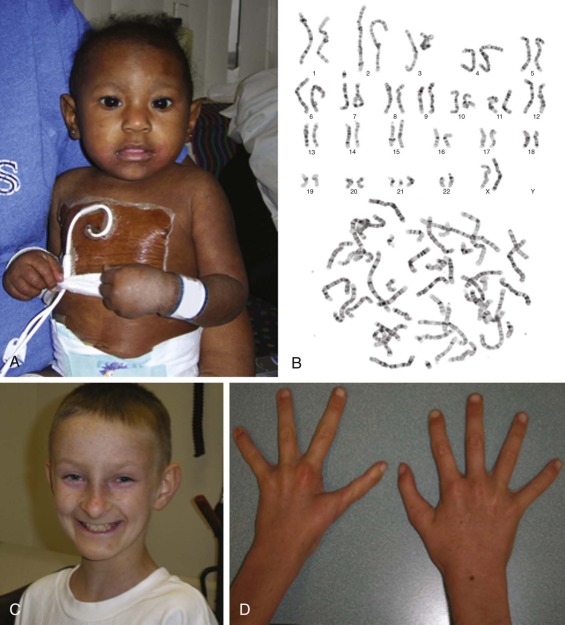
The frequency and nature of congenital abnormalities within the FA population is summarized in Box 7-3 . Figure 7-2, A shows a 1-year-old girl with FA, and Figure 7-2, C shows a 12-year-old boy with several classic phenotypic features, including short stature, microencephaly, broad nasal bridge, epicanthal folds, micrognathia, café-au-lait spots and hypopigmentation, and one absent thumb and one triphalangeal thumb. FA patients may have a combination of the congenital anomalies listed or none of them. The phenotypic variability makes a diagnosis of FA difficult when it is based only on clinical features.
* Abnormalities are listed in the approximate order of frequency within each category.
Skin (40%)
Generalized hyperpigmentation on the trunk, neck, and intertriginous areas; café-au-lait spots; hypopigmented areas
Body (40%)
Short stature, delicate features, small size, underweight
Upper Limbs (35%)
Thumbs (35%): absent or hypoplastic; supernumerary, bifid, or duplicated; rudimentary; short, low-set, attached by a thread; triphalangeal, tubular, stiff, hyperextensible
Radii (7%): absent or hypoplastic (only with abnormal thumbs); absent or weak pulse
Hands (5%): clinodactyly; hypoplastic thenar eminence; six fingers; absent first metacarpal; enlarged, abnormal fingers; short fingers; transverse crease
Ulnae (1%): dysplastic or absent
Lower Limbs (5%)
Feet: toe syndactyly, abnormal toes, flatfeet, short toes, clubfeet, six toes, supernumerary toe
Legs: congenital hip dislocation, Perthes disease, coxa vara, abnormal femur, thigh osteoma, abnormal legs
Gonads
Males (25%): hypogenitalia, undescended testes, hypospadias, abnormal genitalia, absent testis, atrophic testes, azoospermia, phimosis, abnormal urethra, micropenis, delayed development
Females (2%): hypogenitalia; bicornuate uterus; abnormal genitalia; aplasia of uterus and vagina; atresia of uterus, vagina, and ovary
Other Skeletal Anomalies
Head (20%) and face (2%): microcephaly, hydrocephalus, micrognathia, peculiar face, bird-like face, flat head, frontal bossing, scaphocephaly, sloped forehead, choanal atresia, dental abnormalities
Neck (1%): Sprengel deformity; short, low hairline; webbed
Spine (2%): spina bifida (thoracic, lumbar, cervical, occult sacral), scoliosis, abnormal ribs, sacral agenesis, sacrococcygeal sinus, Klippel-Feil syndrome, vertebral anomalies, extra vertebrae
Eyes (20%)
Small eyes, strabismus, epicanthal folds, short or almond-shaped palpebral fissures, hypertelorism, ptosis, slanting, cataracts, astigmatism, blindness, epiphora, nystagmus, proptosis, small iris
Ears (10%)
Deafness (usually conductive); abnormal shape; atresia; dysplasia; low-set, large, or small; infections; abnormal middle ear; absent eardrum; dimples; rotated; canal stenosis
Kidney (20%)
Ectopic or pelvic; abnormal, horseshoe, hypoplastic, or dysplastic; absent; hydronephrosis or hydroureter; infections; duplicated; rotated; reflux; hyperplasia; no function; abnormal artery
Gastrointestinal System (5%)
High-arched palate, atresia (esophagus, duodenum, jejunum), imperforate anus, tracheoesophageal fistula, Meckel diverticulum, umbilical hernia, hypoplastic uvula, abnormal biliary ducts, megacolon, abdominal diastasis, Budd-Chiari syndrome
Urogenital
Males (25%): Micropenis, penile-scrotal fusion, undescended or atrophic or absent testes, hypospadias, chordee, phimosis, azoospermia
Females (2%): bicornate uterus, aplasia or hypoplasia of vagina and uterus, atresia of vagina, hypoplastic uterus, hypoplastic or absent ovary, hypoplastic fused labia
Cardiopulmonary System (6%)
Patent ductus arteriosus, ventricular septal defect, abnormal heart, peripheral pulmonic stenosis, aortic stenosis, coarctation, absent lung lobes, vascular malformation, aortic atheromas, atrial septal defect, tetralogy of Fallot, pseudotruncus, hypoplastic aorta, abnormal pulmonary drainage, double aortic arch, cardiac myopathy
Central Nervous System (3%)
Hyperreflexia, Bell palsy, central nervous system arterial malformation, Moyamoya syndrome, Arnold-Chiari malformation, stenosis of internal carotid artery, small pituitary gland, absent corpus callosum
Slow development (10%)
Endocrinopathies
Data from the International Fanconi Anemia Registry and from the Cincinnati FA cohort indicate that endocrine abnormalities are present in about 80% of children and adults with FA, primarily glucose/insulin abnormalities with a blunted first-phase insulin release, mild hypothyroidism, and male hypogonadism.
Short Stature.
Although short stature is an integral feature of FA, a superimposed endocrinopathy may have an additional impact on growth and is responsive to replacement therapy. Detection of an anatomic abnormality of the hypothalamic-pituitary region by magnetic resonance imaging is a diagnostic marker of permanent growth hormone deficiency. In contrast to previous reports, osteopenia or osteoporosis, when adjusted for body size and height, is not frequent.
Glucose Intolerance.
Impaired glucose tolerance, elevated insulin levels, and overt diabetes mellitus are common in FA. Importantly, HgbA1c or elevated fructosamine does not identify glucose intolerance in FA, possibly because of the high fetal hemoglobin or abnormalities in glycosylation.
Fertility.
Female patients with FA usually have a shortened reproductive life because of secondary amenorrhea, irregular menses, anovulatory menstrual cycles, and premature menopause. Congenital abnormalities in the female genitourinary tract further limit fecundity. Though rare, pregnancies have been reported in women with FA. The babies born were normal, and the pregnancies seemed to have had a transient but not lasting impact on the mother’s hematologic manifestations. Pregnancy in women with FA has also been reported after BMT. Cryptorchidism, hypogenitalism, hypergonadotropic hypogonadism, and reduced sperm counts are frequent in male patients with FA. Reports of male FA patients fathering children are rare.
Hematologic Abnormalities
Progressive BMF is one of the hallmarks of FA, although the hematologic findings vary. Thrombocytopenia and macrocytosis often precede anemia and neutropenia. Elevated hemoglobin F levels and elevated expression of “i” antigen on red cells usually coincide with macrocytosis and are suggestive of stress hematopoiesis. Serum alpha-fetoprotein levels are consistently elevated in FA patients irrespective of the presence of liver abnormalities. Erythropoietin levels are usually elevated in anemic FA patients. The first hematologic abnormalities in individuals with FA are detected at a median age of 7 years. The majority of FA patients already have pancytopenia at the time of diagnosis (53%). By the age of 40, the cumulative incidence of hematologic abnormalities is 90% to 98%.
In rare cases, thrombocytopenia may be present at birth and progress to pancytopenia in the neonatal period or infancy. Thus FA also has to be considered in cases of aplastic anemia in neonates and infants. In other patients, hematologic abnormalities never become clinically evident, and the diagnosis of FA is made later in life as a result of late FA-associated complications such as cancer or leukemia. The emergence of revertant cells leading to hematopoietic mosaicism is often associated with a mild or absent hematologic phenotype (see later). Finally, in a small proportion (2% to 5%) of FA patients, MDS or leukemia is diagnosed but the underlying BMF never becomes clinically apparent. Bone marrow examination generally shows reduced cellularity; however, it may also be normocellular or hypercellular, particularly in disease that evolves into MDS or MDS/acute myeloid leukemia (AML). The cumulative incidence of BMF is approximately 50% by 40 to 50 years of age. BMF is usually progressive. Fifty percent of individuals who are initially found to have thrombocytopenia progress to pancytopenia within 3 to 4 years.
Myelodysplastic Syndrome and Acute Leukemia in Patients with Fanconi Anemia
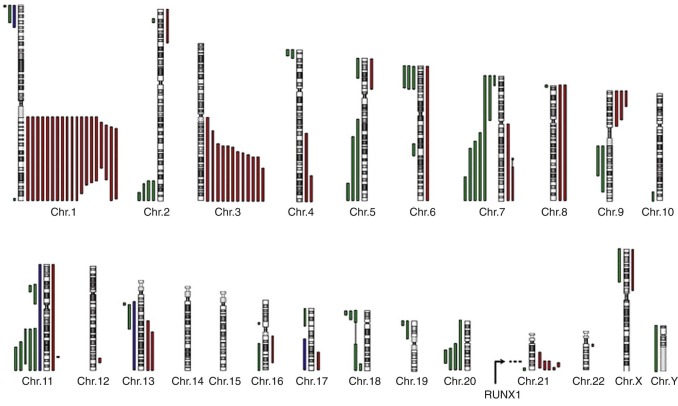
The cumulative incidence of MDS in FA patients is approximately 30%, and of acute leukemia about 10% by the age of 40 to 50 years. In the literature the definition of MDS and acute leukemia in FA patients varies, which makes a strict comparison difficult. Clonal hematopoiesis, as suggested by nonrandom X-inactivation, may be present even before the onset of MDS. Clonal cytogenetic abnormalities are found in 34% to 48% of FA patients. Fluctuations are frequent, including the disappearance of clones, the appearance of new clones, and clonal evolution. Chromosomal abnormalities in MDS most frequently involve chromosomes 1q+, 3q+, −7/7q, 11q− 20q−, 6, 13, and 21q− ( Fig. 7-3 ). Individuals in whom clonal cytogenetic abnormalities develop have a reduced survival rate of 35% to 40% as opposed to 92% in FA patients without such abnormities. An exception is patients with an isolated 1q+ that may be present without morphologic MDS and is not associated with progression to MDS/AML. MDS morphology, as defined by dysplasia in multiple blood cell lineages, portends a poor outcome, with a 5-year survival rate of 9%. Gain of the distal portion of chromosome 3q(26-29) is frequently present in FA patients and is associated with a poor prognosis, decreased survival times, and an increased frequency of MDS/AML.
Leukemia is the most common malignant disease in patients with FA. AML is the most frequent leukemic phenotype, although patients with acute lymphocytic leukemia and chronic myelomonocytic leukemia have been described. Leukemia in patients with FA shows many molecular and clinical similarities to leukemia occurring in other IBMFSs; cases of FA-associated leukemia are notoriously difficult to treat, and most patients die within 6 months of diagnosis. Leukemia in patients with FA may arise de novo or develop from MDS. In some FA patients, AML may be the first hematologic manifestation of disease. Leukemic cells often show complex cytogenetic abnormalities, including abnormalities similar to those seen in MDS. Chromosomal translocations and mutations in oncogenes and tumor-suppressor genes frequently found in de novo AML are rare in FA MDS/AML with the exception of RUNX1 mutations, which are seen in approximately 20% of FA patients with MDS/AML. Abnormalities of chromosome 7 (monosomy, isochromosome, or other structural rearrangement), followed by abnormalities of 1q (usually duplications), are frequent in FA-associated leukemia.
Predisposition to Malignancy
The risk of cancer developing is about 500 to 700 times higher than in the normal population ( Table 7-2 ). By the age of 40 years, the cumulative incidence of solid tumors is estimated to be 26% to 30%. The median age at development of cancer is 16 years, versus 68 years for the same types of cancer in the general population. The risk of solid tumors and leukemia developing increases with age. Thus patients with mild hematologic and phenotypic abnormalities and the longest survival times are at the highest risk for the development of tumors or leukemia. However, with increased survival after hematopoietic stem cell transplantation (HSCT), the frequency of cancer might also increase in patients with more severe disease. The most frequent cancers occur in the neck, head, and upper esophagus, followed by the vulva, anus, and lower esophagus. Human papillomavirus (HPV), particularly HPV16, is frequently isolated from patients with FA. Although the FA DNA repair pathway is involved in limiting HPV replication and patients with FA are frequent carriers of HPV, the role of HPV in the pathogenesis of squamous cell cancer in patients with FA remains controversial. HPV vaccination at an early age should be considered for all patients with FA. The risk for development of squamous cell cancer of the head and neck and particularly the tongue is higher in FA patients after HSCT than in those without HSCT and correlates with the presence and severity of graft-versus-host disease. Adenoma of the liver and hepatocarcinoma were found mainly in patients undergoing androgen therapy. Hepatoadenomas usually regress after cessation of androgen therapy.
| Type | No. of Patients (%) | |||
|---|---|---|---|---|
| Alter et al | Alter et al | Kutler 2003 | Rosenberg | |
| Reporting period | 1927-2001 | 2002-2008 | 1982-2001 | 2001 |
| Patients evaluated | 1301 | 66 | 754 | 145 |
| Hematologic malignant diseases | 214 (16) | 12 (18) | 128 (17) | 32 (22) |
| Cumulative incidence | — | 30 | 33 (by age 40) | 10 (by age 10) |
| Acute myeloid leukemia | 116 (9) | 4 (6) | 60 (8) | 9 (6) |
| Myelodysplastic syndrome | 89 (7) | 8 (12) | 53 (7) | 23 (16) |
| Acute lymphocytic leukemia | 7 | — | 5 | — |
| Chronic myelomonocytic leukemia | — | 1 | 1 | — |
| Lymphoma | 2 | — | 2 | — |
| NONHEMATOLOGIC MALIGNANT DISEASES | 89 (7) | 12 (18) | 73 (10) | 18 (12) |
| Cumulative incidence | — | — | 28 (by age 40) | 29 (by age 49) |
| Squamous cell carcinoma | 66 (5) | 5 (8) | 39 (5) | 16 (11) |
| Oropharyngeal cancer | 26 (2) | — | 19 (3) | 6 (4) |
| Esophageal cancer | 9 | — | 1 | 2 |
| Vulva and anus cancer | 10 | 3 | — | — |
| Vulva cancer | 3 | — | 8 | 3 |
| Anus cancer | 6 | — | 2 | 2 |
| Cervical cancer | 6 | — | 6 | 3 |
| Cutaneous cancer (nonmelanoma) | 6 | 2 | 3 | |
| Hepatic malignant diseases | 37 (3) | — | 18 (2) | 2 (1) |
| Adenoma | 6 | — | 11 | 11 |
| Hepatocellular carcinoma | 14 | — | 6 | 22 |
| Renal cancer | 0 | — | 6 (1) | — |
| Brain cancer | 1 | — | 5 (1) | 1 |
| Miscellaneous tumors | 11 (1) | |||
| Breast cancer | 4 | 1 | 3 | 4 |
| Lung cancer | 3 | — | — | 2 |
| Gastric cancer | 2 | — | — | 2 |
| Colon cancer | 0 | — | — | 1 |
| Osteogenic sarcoma | 1 | — | 1 | 1 |
| Retinoblastoma | 1 | — | — | 1 |
| Other tumors | — | — | 7 | 1 |
The risk and type of cancer vary for different FA subtypes (also see the section on Risk of Malignant Disease). FA patients belonging to complementation group D1 (FA-D1) with biallelic mutations in FANCD1/BRCA2 have a high risk for the development of malignant disease at a very young age, frequently before manifestations of BMF appear, and the cumulative risk for AML or other cancers is 97% by the age of 5.2 years. In addition, the spectrum of cancer is distinct, with medulloblastoma being the predominant malignant disease, possibly because BRCA2 participates in DNA repair outside the FA pathway. An increased risk for childhood cancer with a very similar spectrum has also been found in families belonging to the FA-N subtype. FANCN/PALB2 binds to BRCA2 and facilitates its binding to chromatin. The interaction between these two proteins explains the similar cancer predisposition in FA individuals belonging to the FA-D1 and FA-N subgroups.
All FA cancer patients commonly have a low tolerance for DNA-damaging chemotherapeutic agents. Accordingly, chemotherapeutic regimens often need to be modified, given at low dosage, or avoided in favor of alternative or surgical approaches.
Diagnosis
The most widely used diagnostic test for FA is hypersensitivity to the clastogenic (chromosome-breaking) effect of diepoxybutane (DEB) or mitomycin C (MMC). The increased spontaneous chromosomal instability of FA cells leads to distinctive chromosomal breaks and gaps and various chromatid interchanges, which were previously used as a cellular marker for FA. The frequency of spontaneous chromosomal instability in FA cells is highly variable, and it is thought to be responsible for the increased susceptibility of FA patients to cancer. The sensitivity of FA cells to the clastogenic effect has been studied for numerous cross-linking agents, but DEB and MMC are the agents most widely used for the diagnosis of FA. Figure 7-2, B shows the increased spontaneous chromosome fragility in peripheral lymphocytes in a patient with FA. The diagnosis of FA is made when DEB-treated cultured lymphocytes demonstrate a 3- to 10-fold increase in chromosomal breakage over normal controls. The tests are highly sensitive and fairly specific for FA and are used for the diagnosis of FA before the development of clinical disease and for prenatal diagnosis. Possible rare exceptions that can result in a pattern of DEB- or MMC-induced chromosomal breakage similar to that found in FA patients are individuals with specific mutations in the NBS1 gene in Nijmegen breakage syndrome and patients with cohesinopathies, that is, those with Roberts syndrome and Warsaw breakage syndrome. However, more frequently, the presence of revertant cells in the specimen may complicate the interpretation of DEB or MMC test results (see the section on Somatic Mosaicism, Functional Reversion, and Attenuation of Fanconi Anemia Cells). It is estimated that 10% to 25% of FA patients exhibit two populations of phytohemagglutinin-stimulated lymphocytes, one that is sensitive to the clastogenic effects of DNA cross-linking agents and one that is resistant to them. Thus the presence of only a few metaphases with multiple structural chromosomal changes might indicate FA with somatic mosaicism. In these cases, demonstration of increased chromosomal breakage in DEB- or MMC-treated fibroblasts might help confirm the diagnosis of FA.
Immunoblotting for FANCD2 Monoubiquitination
Biallelic mutations in 10 of the 15 FA genes ( FANC genes) lead to failure of FANCD2 monoubiquitination, and failure of FANCD2 monoubiquitination is specific for a defect in the FA pathway (see the section on Function of FANC Proteins). Testing for FANCD2 monoubiquitination by immunoblotting of peripheral blood lymphocytes and fibroblasts is therefore used in some laboratories as an alternative first-line tool for the diagnosis of FA. Biallelic mutations in FANCD1/BRCA2 , FANCJ , FANCN/PALB2 , FANCO/RAD51C, and FANCP/SLX4 that have normal FANCD2 monoubiquitination but an abnormal DEB or MMC test (or both) are missed with this assay and, therefore, additional testing is required.
Genetic Testing for FANC Gene Mutations
Only the identification of biallelic mutations in a FANC gene definitively confirms the diagnosis of FA. Currently genetic testing for FANC gene mutations is not usually recommended as a first-line tool for the diagnosis of FA. Possible exceptions are families with a known FANC gene mutation and FA in a population with a high frequency of a founder mutation, for example, a case of FA in Spanish gypsies. The indications, advantages, and difficulties of genetic testing for biallelic mutations in FANC genes are discussed later. As costs decrease, targeted or whole-genome next-generation sequencing is likely to become the diagnostic test of choice for patients clinically suspected to have FA (see the section on Molecular Diagnosis of Fanconi Anemia).
Cellular Phenotype of Fanconi Anemia Cells
In addition to increased spontaneous and induced chromosomal instability, FA cells have several other phenotypic characteristics ( Table 7-3 ). FA cells have increased sensitivity to other DNA-damaging agents, such as ionizing radiation and oxygen radicals. FA cells have an abnormality in cell cycle distribution, with an increased number of cells with 4N DNA content, suggesting a delay in the G 2 /M or late S phase (or both) of the cell cycle. FA cells have a failure in cytokinesis forming an increased number of ultrafine DNA bridges (UFB) during the M phase of the cell cycle, leading to increased chromosome instability and binucleated cells that undergo apoptosis. The increase in cells with 4N DNA content is accentuated after treatment with interstrand DNA cross-linking agents. Flow cytometric measurements of the increase in the proportion of cells with 4N DNA content after such treatment may be used as an additional criterion for the diagnosis of FA. However, similar results have been obtained in patients with ataxia-telangiectasia. FA cells show accelerated telomere shortening in vitro and an increase in telomere free ends and chromosome end fusions, suggesting a defect in maintenance of telomere integrity and increased breakage at telomeric sequences. Cells deficient in the FA repair pathway are hypersensitive to both formaldehyde and acetaldehyde.
| Feature | References |
|---|---|
| Spontaneous chromosome breaks | |
| Sensitivity to cross-linking agents | |
| Sensitivity to formaldehyde | |
| Prolongation of G 2 /M phase and S phase of cell cycle | |
| Failure in cytokinesis, with increased number of ultrafine DNA bridges (UFB) during M phase, binucleated cells | |
| Accelerated telomere shortening | * |
| Hypersensitivity to ionizing radiation † | |
| SENSITIVITY TO OXYGEN | Reviewed in |
| Poor growth at ambient O 2 | |
| Overproduction of O 2 radicals | |
| Deficient O 2 radical defense | |
| Deficiency in superoxide dismutase | |
| Overproduction of TNF-α | |
| Increased TNF-α and IFN-γ sensititivy | |
| DIRECT DEFECTS IN DNA REPAIR | |
| Defective repair of DNA cross-links | |
| Hypermutability (by deletion) | |
| Increased apoptosis | |
| Increased induction of p53 | |
| Defective end-joining fidelity | |
| Increased homologous recombination | |
| INTRINSIC STEM CELL DEFECT | |
| Decreased colony growth in vitro | |
| Decreased gonadal stem cell survival |
* Experiments in mice suggest that accelerated telomere shorenting is not a direct effect of FA gene products on telomeres but rather an indirect effect, such as the increased sensitivity to oxygen radicals.
† Hypersensitivity to ionizing irradiation is controversial. More recent results suggest an increased sensitivity in the majority of fa complementation groups.
Differential Diagnosis
Clinically, FA has to be differentiated from other IBMFSs, particularly dyskeratosis congenita (DC), Diamond-Blackfan anemia (DBA), Shwachman-Diamond syndrome (SDS), Wiskott-Aldrich syndrome (WAS), thrombocytopenia with absent radii (TAR), and other nonclassified forms of BMF. Clinical manifestations, associated physical abnormalities, family history (pattern of inheritance), and increased chromosomal fragility in the DEB or MMC test performed on cultured peripheral lymphocytes or fibroblasts, or both, are helpful in strengthening the diagnosis of FA. However, only identification of pathogenic mutations on both copies of an FA gene (biallelic mutation) may definitely consolidate the diagnosis of FA. Congenital abnormities in FA may have an overlap with malformations seen in other disorders with genetic and nongenetic causes. Thrombocytopenia and radial ray abnormalities are also characteristic of TAR. However, in FA, if the radii are affected, the thumbs are always abnormal; in TAR, in which the radii are absent, thumbs are always present. Other syndromes that have phenotypic overlap with FA include Holt-Oram syndrome (MIM 142900), which is characterized by thumb anomalies and atrial septal defects and caused by mutations in the TBX5 gene, and Baller-Gerold syndrome (MIM 218600) and Rothmund-Thomson syndrome (MIM 278400), which are caused by mutations in the RECQL4 gene and characterized by radial defects, craniosynostosis, and cancer predisposition but not BMF. FA patients may present with VATER (vertebral defects, imperforate anus, tracheoesophageal fistula, and radial and renal dysplasia) or VACTERL (vertebral abnormalities, anal atresia, cardiac abnormalities, tracheoesophageal fistula and/or esophageal atresia, renal agenesis and dysplasia, and limb defects) (MIM 192350) or the IVIC syndrome (MIM 147750), which is an acronym for Instituto Venezolano de Investigaciones Cientificas, the institution at which a family with autosomal dominant radial ray defects, hearing impairment, internal ophthalmoplegia, and thrombocytopenia was first described. In patients presenting with these associated anomalies, FA should be excluded.
The increased chromosomal breakage in FA has to be distinguished from that seen in Bloom syndrome (MIM 210900), another single-gene disorder with a predisposition for leukemia as a result of mutation in the helicase RecQ protein-like-3 gene ( RECQL3 ). Chromosomal aberrations are in general more numerous in FA cells than in Bloom cells. Furthermore, in Bloom syndrome, most interchanges occur between homologous chromosomes, whereas in FA they are more frequently seen between nonhomologous chromosomes.
Nijmegen breakage syndrome (NBS; MIM 251260), caused by mutations in the NBS1 gene and characterized by immunodeficiency and a predisposition to lymphoma, is another autosomal recessive chromosomal breakage disease. A small number of patients with NBS may have FA-like features, including BMF and the development of myeloid leukemia, and specific mutations in the NSB1 gene cause an increased clastogenic response to MMC and DEB testing.
A third disease associated with spontaneous chromosomal breakage and hematopoietic malignant disease is ataxia-telangiectasia (MIM 208900), which is inherited in an autosomal recessive manner. Cells from patients with ataxia-telangiectasia are hypersensitive to ionizing radiation but not to cross-linking agents. Furthermore, the disease is associated with immunodeficiency and progressive cerebellar neuronal degeneration, which clearly distinguish this condition from FA.
Seckel syndrome ( SCKL1 , MIM 210600), caused by a defect in the signaling pathway dependent on ataxia-telangiectasia and Rad3-related (ATR), may have some clinical similarities to FA, including anemia and an increased risk for AML.
Patients deficient in DNA ligase IV ( LIG4, MIM 606593) are characterized by microcephaly, growth retardation starting in utero, distinctive facial appearance (birdlike face), developmental delay, immunodeficiency, pancytopenia, and pronounced clinical and cellular radiosensitivity. LIG4 deficiency is another rare IBMFS caused by defective DNA repair.
Two of the cohesinopathies, Roberts syndrome (MIM 268300) (caused by mutations in ESCO2, encoding an acetyltransferase involved in sister chromatid cohesion) and Warsaw breakage syndrome (MIM 613398) (caused by mutations in DDX11/ChlR1, encoding an XPD-like DNA helicase), show excessive MMC-induced chromosomal breakage in primary lymphocyte cultures and an increased arrest of primary skin fibroblasts in the G 2 /M phase of the cell cycle that need to be distinguished from FA.
Genetic Characteristics
Inheritance.
FA is a genetically heterogeneous recessive disorder. Mutations in at least 15 different genes, all of which have been identified, may be responsible for FA. Inheritance of FA is autosomal or, in rare cases, X-linked.
Complementation Groups.
At least 16 genetic subtypes or complementation groups have been identified (FA-A, FA-B, etc.), each with a distinctive disease gene (Fanconi anemia gene A [ FANCA ], FANCB , etc.). The majority of patients belong to complementation group A (≈ 60% to 70%), followed by complementation groups C and G. Table 7-4 shows the complementation groups and the genes responsible for FA and their frequency. Complementation groups were originally defined by somatic cell fusion and the ability of the heterokaryon to correct the MMC hypersensitivity as measured in a growth inhibition assay. Correction of the MMC hypersensitivity indicates that the two fusion partners belong to different complementation groups, whereas no correction indicates that they belong to the same complementation group. More recently and as more FANC genes are identified, complementation groups are assigned through the correction of MMC hypersensitivity after introduction of the respective wild-type cDNA (complementation by cDNA expression). Retroviruses expressing FANC cDNA may be used to correct the phenotype of T cells from FA patients, which determines the complementation group in a rapid, accurate manner.
| Complementation Group | Responsible Gene | Percentage of FA Patients | Locus | Protein (kDa) | FANCD2 Monoubiquitination | Main Function of Protein | Comments | References |
|---|---|---|---|---|---|---|---|---|
| FA-A | FANCA | ≈66 | 16q24.3 | 163 | + | FA core complex | Phosphorylated following DNA damage | |
| FA-B | FANCB | ≈2 | Xp22.31 | 95 | + | FA core complex | ||
| FA-C | FANCC | ≈10-15 | 9q22.3 | 63 | + | FA core complex | ||
| FA-D1 | FANCD1/BRCA2 | 3-4 | 13q12-13 | 380 | − | Recruits RAD51 and promotes HR repair | ||
| FA-D2 | FANCD2 | ≈3 | 3p25.3 | 155, 162 | + | Monoubiquitinates and recruits FANCP to chromatin | Phosphorylated and monoubiquitinated following DNA damage | |
| FA-E | FANCE | 2-3 | 6p21.3 | 60 | + | FA core complex, probable adaptor for FANCD2 | Direct binding to FANCD2, phosphorylated following DNA damage | |
| FA-F | FANCF | ≈2 | 11p15 | 42 | + | FA core complex | ||
| FA-G | FANCG/XRCC9 | 8-10 | 9p13 | 70 | + | FA core complex | ||
| FA-I | FANCI | <2 | 15q25-26 | 150 | + | Monoubiquitinated protein (part of FANCI-FAND2 subcomplex) | Phosphorylated and monoubiquitinated following DNA damage, binds to FACND2 | |
| FA-J | FANCJ/BACH1/Brip1 | <2 | 17q22.3 | 150 | − | 5′→3′ DNA helicase/ATPase | Binding to BRCA1 | |
| FA-L | FANCL/PHF9/POG/FAAP43 | Rare | 2p16.1 | 43 | + | FA core complex, ubiquitin ligase | Recruits UBE2T, monoubiquitinates FANCD2 and FANCI | |
| FA-M | FANCM/ Hef/ FAAP250 | Rare | 14q21.3 | 250 | + | FA core complex, branch migration/ATPase activity, sensor for DNA cross-link damage | Helicase/nuclease motif, phosphorylated following DNA damage, interacts with BLM | |
| FA-N | FANCN/PALB2 | Rare | 16p12.1 | 130 | − | Mediates interaction between BRCA1 and BRCA2 during HR | Binding to BRCA2 | |
| FA-O | FANCO (RAD51C) | Rare | 17q22 | 43 | − | Promotes HR, interacts with RAD51 and RAD51 homologues | ||
| FA-P | FANCP (SLX4) | Rare | 16p13.3 | 200 | − | Functions as holiday junction resolvase with SLX1 | Interacts with XPF-ERCC1 and EME1-MUS81 nucleases |
Fanconi Genes ( FANC Genes).
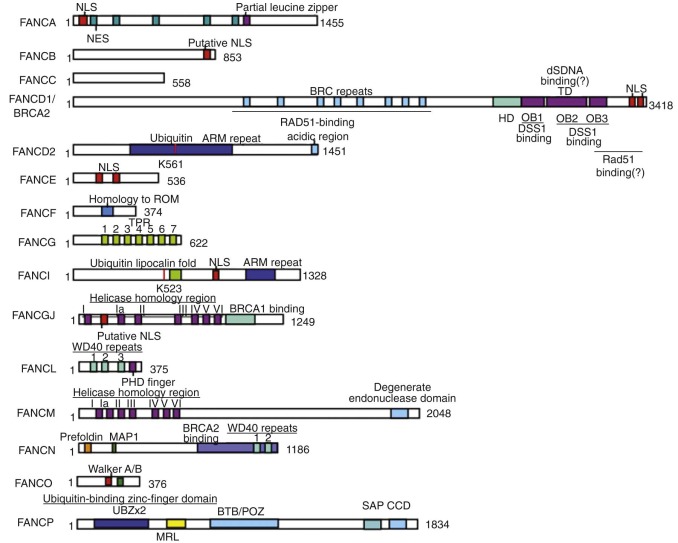
FANC genes have been identified by expression cloning, positional cloning, protein complex purification, or a combination thereof and are summarized in Table 7-4 . Figure 7-4 schematically represents the 15 FANC genes and their functional domains.
Function of FANC Proteins.
The FANC proteins have been shown to act mainly during the S phase of the cell cycle. Their main role is in the repair of DNA interstrand cross-links that impede replication and transcription by inhibiting DNA strand separation. It is generally thought that FANC proteins sense, promote, and coordinate three DNA repair pathways: Repair is initiated by recognition of a lesion by nucleotide excision repair components, followed by incisions made flanking the interstrand cross-links; the gapped intermediate that is produced is filled by translesion synthesis or by repair of broken replication forks by homologous recombination. Thus it is thought, and schematically summarized in Figure 7-5 , that in normal cells the FA pathway serves to minimize the severity of mutational outcomes by favoring error-free DNA repair and error-prone DNA repair that results in base pair substitutions or small deletions over DNA repair that results in larger deletions and chromosomal rearrangements ( Box 7-4 summarizes the main DNA repair pathways).
DNA repair pathways are highly regulated pathways that involve detection of DNA lesions, signaling to recruit DNA repair factors, activation of DNA repair enzymes, and disassembly or degradation of DNA repair factors after repair.
- •
EXCISION OF DAMAGED AND MISMATCHED BASES
- •
Nucleotide excision repair, including global genome repair and transcription-coupled repair
- •
Base pair excision repair
- •
Mismatch repair
- •
- •
REPAIR OF DOUBLE-STRANDED BREAKS
- •
Homologous recombination (generally error free)
- •
Nonhomologous end joining (generally error prone)
- •
- •
DNA STRESS REPLICATION PATHWAY, REPLICATION PATHWAY OF DAMAGED TEMPLATES DURING DNA SYNTHESIS
- •
Translesion DNA synthesis (error prone)
- •
The current model suggests that FANCM recognizes the DNA interstrand cross-links and initiates DNA repair by recruiting eight FA core complex proteins (FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL, and FANCM) to the DNA damage site, stabilizing the stalled replication fork, and initiating ATR mediated checkpoint signaling. The FA core complex functions as an ubiquitin ligase, of which FANCL is the catalytic subunit. FANCE recruits FANCD2 to the FA core complex, whereas disease-associated FANCE mutations disrupt the FANCE-FANCD2 interaction. Figure 7-6 schematically summarizes the current view of the FANC protein interconnections in the FA DNA repair pathway. During activation, multiple FA proteins undergo phosphorylation by ATR-CHK1 checkpoint kinases, highlighting the close interconnection of the FA pathway with DNA damage response signaling. The FA core complex catalyzes monoubiquitination of the FANCI-FANCD2 complex, which is a key regulator of the FA pathway. A defect in any of these core proteins results in failure to monoubiquitinate the FANCI-FANCD2 complex, underscoring the importance of this modification in the FA pathway. FANCI and FANCD2 are dependent on each other for their respective monoubiquitination. The FANCL subunit is a ubiquitin E3 ligase, and in concert with the UBE2T E2 enzyme, it conjugates a single ubiquitin to FANCD2 and to FANCI. The monoubiquitinated FANCI-FANCD2 complex is relocalized to the DNA lesion, where it coordinates cross-link repair together with downstream FA proteins. Monoubiquitinated FANCD2 acts as a platform to recruit multiple nucleases, including FANCP, to coordinate nucleolytic incisions flanking the interstrand cross-links. The incision creates a DSB, which is repaired by homologous recombination. Downstream FA proteins, including FANCD1, FANCJ, FANCN, and FANCO, promote RAD51-dependent strand invasion and the resolution of homologous recombination intermediates.
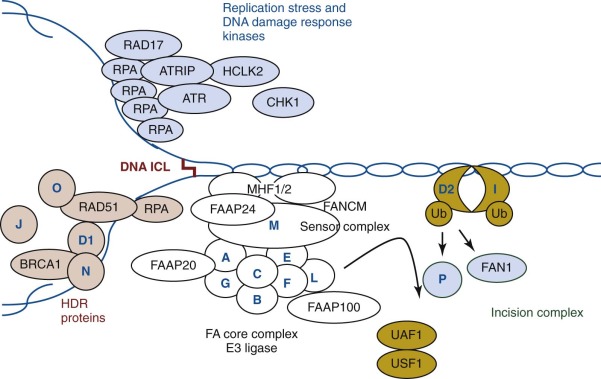
The FA core complex is assembled sequentially from subcomplexes, which may have additional functions aside from DNA repair. Several FA proteins, including FANCA, FANCE, FANCG, FANCD2, FANCI, and FANCM, are posttranslationally modified by phosphorylation, and two FA proteins, FANCC and FANCD2, are regulated through a caspase-mediated proteolytic process. FANC proteins have been reported to interact with a number of other proteins, including proteins involved in transcription, the cell cycle, oxidative metabolism, cell signaling, and cytokine response, suggesting that FANC proteins/subcomplexes might have a separate role in cell cycle control, apoptosis, hematopoiesis, and tumorigenesis that is independent of DNA damage recognition/repair.
Bone Marrow Failure in Fanconi Anemia
Although the impairment of DNA repair seems to explain the genomic instability and possibly the increased risk for malignant disease in patients with FA, its link to the pathogenesis of BMF is less obvious, because mutations associated with genomic instability outside the FA DNA repair pathway are not usually associated with BMF. There is an ongoing debate on whether BMF is a direct consequence of the FA DNA repair defect or is caused by impairment of functions apart from DNA repair. Naturally, FA DNA repair and non–DNA repair pathways in the pathogenesis of BMF may not be mutually exclusive but, rather, may overlap. However, two recent findings point strongly towards defective DNA repair being at least the initiating cause of BMF in FA. In a recent study Ceccaldi and colleagues demonstrated that the basal and MMC-induced levels of p53 and its downstream target p21 are elevated in the blood, bone marrow, and liver of fetuses with FA, with a simultaneous accumulation of cells in phase G 0 /G 1 . These findings suggest that in FA patients the pool of available hematopoietic stem cells and progenitor cells is compromised, with fewer cells and less proliferation of cells than in normal individuals. The impairment is intrinsic, occurs even in the fetus, and leads to overt BMF later in life. Although cross-linking agents have been used experimentally to demonstrate the FA defect in DNA repair, experiments by Ridpath and colleagues and Langevin and colleagues suggest that naturally occurring aldehydes might be a major source of endogenous cross-linking and FA pathway activation under steady-state conditions in vivo. Interestingly, HSCs express aldehyde hydrogenase type 2 ( Aldh2 ), a condition that is essential for the protection of HSCs against acetaldehyde toxicity. The experiments were performed in mice and need to be confirmed in patients with FA, but the findings suggest that the FA pathway is needed to protect HSCs from aldehyde-mediated genotoxicity (see the section on Mouse Models for Fanconi Anemia). Moreover, these observations explain the tissue specificity of HSC and solve the conundrum of why HSC is affected in FA but is not affected, or not to the same extent, in other non-FA DNA repair disorders.
FANC Gene Mutations in Fanconi Anemia
Detailed information on mutations identified in the FANC genes in patients with FA is maintained in the FA Mutation Database at Rockefeller University through the International Fanconi Anemia Registry (available at rockefeller.edu/fanconi/ ) and in the Leiden Open Variation Database; chromium.liacs.nl/lovd/ ). Frequent FA mutations and mutations with founder effects are presented in Table 7-5 .
| Gene | Founder Effect | Variation at DNALevel (Old Nomenclature) * | Variation at Protein Level * | Frequency within Mutated FA Allele | Population with Founder Effect (% of All FA Patients) | References |
|---|---|---|---|---|---|---|
| FANCA | Yes | c.3788_3790delTCT † | p.1263delF | 8.8% | 51% Brazilians, relatively mild | |
| No | c.1115-1118delTTGG † | p.V372fs | 5.5% | 2.2% Brazilians | ||
| Yes | Intragenic deletion of exons 12-31, intragenic deletion of exons 17-31 | del | 60% Afrikaner in South Africa | |||
| Yes | c.295C→T | p.Q55X | Spanish Gypsies (highest prevalence of mutant FANCA allele) | |||
| Yes | c.65G→A | W22X | Unique to Ashkenazi Jews | |||
| FANCC | Yes | c.711 +4A→T (IVS4+4A→T) | Splicing | Homozygous in 80% of Ashkenazi-Jewish FA (carrier frequency 1/100), relatively common in Japan; severe phenotype in Jews, milder in Japanese | ||
| Yes | c.67delG (322delG) | fs | Homozygous in 50% of Dutch FA patients, relatively mild phenotype | |||
| FANCG | Yes | c.1077-2A→G | Splicing | Portuguese Brazilians | ||
| Yes | c.1480+1G→C | Splicing | French Acadians | |||
| Yes | c.307+1G→C | Splicing | Koreans and Japanese | |||
| Yes | c.1794_1803del10 | p.W599fs | Europeans | |||
| Yes | 313G→T | E105X | 44% of mutated FANG alleles in Germans | |||
| Yes | c.637 643del | p.Y213fs | Sub-Saharan Africa, 82% of all black FA patients | |||
| FANCD1/BRCA2 | Yes | c.6174delT | fs | Unique to Ashkenazi-Jewish persons | ||
| No | c.859+1G→A (IVS7+1G→A) ‡ | Splicing | ||||
| No | c.859+1G→A (IVS7+1G→A) ‡ | Splicing | ||||
| No | c.886delGT § | fs | ||||
| No | c.6174delT § | fs | ||||
| FANCD2 | Yes | c.1948-16T → G | Splicing | Turkish | Founder mutation | |
| FANCJ | c.2392C → T | p.R798X | 50% | Found in ≈50% of FA-J patients of diverse ancestry; ancient mutation or hot spot |
* For coding DNA reference sequence see rockefeller.edu/fanconi/, with the first base of the Met-codon counted as position 1. Single-letter abbreviation is used for amino acids: E , glutamic acid; F , phenylalanine; Q , glutamine; R , arginine; W , tryptophan; X , stop codon; Y , tyrosine.
† Most frequent FANCA gene mutations.
‡ Associated with high frequency of leukemia at very young age (acute myeloid leukemia and acute lymphocytic leukemia).
FANCA is the gene most frequently mutated in FA patients. The mutations are spread throughout the gene and include missense and nonsense mutations, small deletions, insertions, and duplications, as well as splicing mutations. Of note is the substantial number of large genomic, usually intragenic DNA deletions, which account for about 40% of all pathogenic mutations within the FANCA gene and are thought to be due to the frequent occurrence of Alu repeats in this genomic region. Large gene deletions are not generally detected by routine nucleotide sequence–based mutation detection methods but require more specialized analysis (haplotype analysis) or molecular methods specifically designed for the detection of large gene deletions. Two small deletions (deletion of three nucleotides between positions 3788 and 3790, with c.3788_3790delTCT leading to deletion of phenylalanine at position 1263, 1263delF, and a 4–base pair (bp) deletion at position 1115, c.1115-1118delTTGG, leading to a frameshift and premature stop codon) are the most common pathogenic mutations in the FANCA gene, with a frequency of 8.8% and 5.5% in the non-Brazilian and 51.1% and 2.2% in the Brazilian mutated FANCA alleles (see Table 7-5 ).
Mutations in FANCC genes account for 10% to 15% of all FA cases. The splicing mutation c.711+4A→T (old nomenclature, IVS4+4A→T) in the FANCC gene accounts for the majority (80%) of FA cases in Ashkenazi Jewish families. Mutations in FANCG are responsible for about 8% to 9% of patients with FA. Several mutations with founder effects have been identified in specific populations (see Table 7-5 ).
Biallelic mutations in BRCA2 are found in individuals with FA-D1. Biallelic FANCD1 / BRCA2 mutations account for about 3% to 4% of all FA patients. The majority of FANCD1 / BRCA2 mutations in FA lead to frameshifts or truncations (see Table 7-5 ). Interestingly, however, FA-D1 cells are not biallelic for FANCD1 / BRCA2 null alleles but have at least one hypomorphic FANCD1 / BRCA2 allele expressing a FANCD1/BRCA2 protein with some residual activity. Biallelic null FANCD1 / BRCA2 mutations have never been identified and in mice are embryonically lethal. Biallelic mutations in FANCD1 / BRCA2 have a very high risk of early malignant disease. The splice-site mutations c.859+1G→A (IVS7+1G→A) and c.859+1G→A (IVS7+1G→A) are associated with a high frequency of leukemia at a very young age, whereas the frameshift mutations c.886delGT and c.6174delT are associated with brain tumors.
Mutation in FANCD2 accounts for 3% to 6% of FA-affected patients. Malformations are frequent in these patients, and hematologic manifestations appear early and progress rapidly. Although hypomorphic mutations exist, patients with FANCD2 mutations have a relatively severe form of FA. Mutation analysis revealed 66 mutated alleles, 34 causing aberrant splicing. Many mutations are recurrent and have ethnic associations and shared allelic haplotypes. There were no biallelic null mutations, suggesting that in contrast to the Fancd2 knockout mouse, complete absence of FANCD2 does not exist in patients with FANCD2 mutations.
Mutations in FANCN/PALB2 were identified in families with the new subtype FA-N and cancer in early childhood. Similar to biallelic BRCA2 mutations, biallelic FANCN/PALB2 mutations confer a high risk for childhood cancer.
Molecular Diagnosis of Fanconi Anemia
Molecular diagnosis is not routinely performed for FA, because it is expensive, labor intensive, and, in some cases, possible only in specialized laboratories using research tests that are currently not approved by Clinical Laboratory Improvement Amendments. However, only identification of biallelic mutations in a FANC gene (or hemizygosity for a mutation in the FANCB gene) definitively establishes the diagnosis of FA. Furthermore, molecular diagnostic testing enables the identification of mutation carriers, facilitates prenatal diagnosis for an existing pregnancy, and allows preimplantation genetic diagnosis for partners identified as mutation carriers. For a definition of preimplantation genetic diagnosis see Box 7-5 . In addition, because the clinical manifestations and severity of disease may vary depending on the affected FANC gene and the nature of individual mutations, molecular diagnostic testing may influence medical management not only for FA individuals carrying biallelic FANC gene mutations but possibly also for heterozygous FANC gene mutation carriers (see the section on Heterzygous Carriers of FANC Gene Mutations). Currently, an educated candidate gene approach is used to identify the affected FANC gene by taking into account the origin and ethnicity of the affected FA individual. After exclusion of the most frequent mutations, it might be necessary to determine the complementation group and perform mutational screening of the entire coding sequence, including methods that will detect large deletions in compound heterozygotes. Protein expression analysis and functional assays are used to test for the pathogenicity of unclassified variants. Multiplexed next-generation sequencing with massively parallel sequencing is becoming increasingly available for clinical genetic testing and has been shown to be an effective molecular diagnostics approach for FA. With the decreasing costs of next-generation sequencing, customized targeted, whole-exome, or whole-genome sequencing is likely to become the future diagnostic standard for patients with FA, enabling genetic molecular diagnosis to be performed at a reasonable price in a short turnaround time ( Box 7-6 ). Although genetic mutation analysis is likely to be successful in the majority of FA patients, in some, the responsible gene mutations might remain unclear. In addition to the large number of mutations in FANC genes associated with FA, a number of nonpathogenic mutations and mutations of unknown pathogenic significance are identified and require further functional investigations. The multitude of FA complementation groups, the presence of pseudogenes, the heterogeneity of the mutation spectrum, and the frequency of large intragenic deletions present a considerable challenge for the current and future molecular diagnosis of FA.
Preimplantation genetic diagnosis (PGD) is the genetic diagnosis of a preimplantion embryo (typically on day 3 of embryo development at the 8-cell stage or on day 5 as a blastocyst) obtained by in vitro fertilization. It is usually performed in families with a severe genetic disorder or for selection of a human leukocyte antigen–compatible donor for a sibling in need of a hematopoietic stem cell transplant. The procedure is often successful but is ethically controversial.
DNA sequencing determines the order of the nucleotide bases (adenine, guanine, cytosine, thymine) in a molecule of DNA. Next-generation sequencing is a high-throughput technology that uses massively parallel sequencing, producing thousands or millions of sequences at once. Next-generation sequencing reduces the turnaround time and number of assays and significantly reduces the cost of analysis. In the future, next-generation sequencing will allow simultaneous screening for mutations in most or all of the loci associated with inherited bone marrow failure syndromes at once as well as screen the whole genome for the detection of novel mutations. With decreasing costs, next-generation sequencing is likely to become the diagnostic test of choice for all patients clinically suspected to have an inherited bone marrow failure syndrome or presenting with bone marrow failure in general.
Heterozygous Carriers of FANC Gene Mutations
Heterozygous carriers of FANC gene mutations are not at risk for the development of BMF. Similarly, cell lines heterozygous for FANC gene mutations do not show the chromosomal instability (either spontaneously or in response to cross-linking agents) characteristic of FA cells with biallelic FANC gene mutations. However, an increased frequency of skeletal and genitourinary abnormalities and an increased risk of malignant disease have been reported in heterozygous FANC D1/J/N/O and possibly FANCP and female FANCB gene mutation carriers.
Risk of Malignant Disease in Heterozygous Carriers.
A strong association exists between heterozygous mutation of FANC genes and susceptibility to breast or ovarian cancer, and other forms of malignant disease have been identified in heterozygous mutation carriers. Carriers of mutations in BRCA1 or BRCA2/FANCD1 have a 65% and 35% risk, respectively, of developing breast cancer and a 39% ( BRCA1 ) and 6% ( BRCA2/FANCD1 ) risk for ovarian cancer. Heterozygous BRCA2/FANCD1 mutations additionally predispose to pancreatic, prostate, and gastric cancers, as well as melanoma. Monoallelic truncating FANCN/PALB2 mutations are associated with familial breast cancer, although the risk of breast cancer is lower (2.3-fold) than that in BRCA2 mutation carriers. Heterozygosity for FANCJ mutations has been identified in patients with early-onset breast cancer. Monoallelic pathogenic mutations in RAD51C/FANCO confer an increased risk for breast and ovarian cancers. Heterozygous germline mutations in FANCC and FANCG and loss of heterozygosity or functional biallelic loss in the tumor sample have been identified in rare patients with pancreatic cancer. Furthermore, heterozygous germline mutations of FANCA have been identified in a small percentage of patients with AML.
Somatic FANC Gene Mutations in Cancer
Cancer cells often accumulate a large number of mutations within an individual cancer cell, many of which promote cell growth and survival. It is therefore thought that a cancer cell with an increased mutation rate, a mutator phenotype, will have a selective advantage in tumor growth and development. Inactivation or mutation of genes involved in maintaining genetic stability is thought to be an early event in cancer development. The FA pathway has been observed to be one of the pathways that maintains genomic stability and is targeted in several forms of sporadic cancer.
Identification of acquired abnormalities in the FA pathway might become clinically important because these tumors are expected to have increased sensitivity to cross-linking agents such as cisplatin, cyclophosphamide, MMC, and other novel agents. Thus identification of abnormalities in the FA pathway may serve as a biomarker that identifies a subgroup of patients with cancers selectively sensitive to specific chemotherapeutic agents. Similarly, inhibition of the FA pathway in cancer may render cancer cells sensitive to chemotherapy.
Genotype-Phenotype Correlation
Clinical findings in FA are highly variable, although within an individual family the presence or absence of multiple congenital abnormalities or the age at onset of other disease manifestations is usually concordant. More recent studies have demonstrated that disease severity and manifestations are determined, at least in part, by the FANC gene affected (complementation group) and by the nature of the mutations. For example, patients with mutations in the FANCA gene usually have a milder form of disease and BMF develops at a later age than it does in FA individuals with mutations in the FANCC or FANCG gene, whereas in FA patients with biallelic FANCD1 or FANCN mutations, cancer develops even before the manifestation of BMF. Furthermore, splice-site mutations in FANCD1 are more frequently associated with the development of leukemia, whereas frameshift mutations within the same gene more frequently lead to brain tumors (see the section on FANC Gene Mutations in Fanconi Anemia). Malformations are frequent in FANCD2 patients, and hematologic manifestations appear earlier and progress more rapidly than they do in non- FANCD2 patients.
FA-B is X-linked, in contrast to all other forms of FA, which are autosomal. Thus a single mutation is sufficient to cause a phenotype and disease in males. In females, because of inactivation of one of the two X chromosomes, a phenotype is expressed only in cells with the FANCB mutation on the active X chromosome. Because X-chromosome inactivation occurs at random in early embryogenesis and, once determined, persists clonally, one might expect female FANCB mutation carriers to have a mild form of FA-B. Interestingly, however, to date no female FA-B patient with clinical manifestations of FA has been reported, and careful clinical examination of females heterozygous for FANCB gene mutations showed no manifestations of disease. Molecular studies investigating X-inactivation revealed that only the X chromosome with the normal FANCB gene is expressed in blood and bone marrow and also in fibroblasts. This finding indicates a proliferative disadvantage of cells expressing the mutant FANCB allele and complete outgrowth by phenotypically normal cells, not only in bone marrow but possibly also in all other tissues. This situation differs from other X-linked IBMFSs such as X-linked DC (see the section on Dyskeratosis Congenita). For individuals carrying a FANCB mutation, however, longer follow-up and examination of additional mutation carriers will be necessary to determine whether some FA-B cells persist in female carriers and whether these persisting FA-B cells are at risk for malignant transformation. Whether somatic FANCB inactivation can be found in malignant disease (only a single mutation on the active X chromosome is required) remains to be determined.
Somatic Mosaicism, Functional Reversion, and Attenuation of Fanconi Anemia Cells
About 10% to 25% of FA patients have been found to have mosaicism in the peripheral blood inasmuch as two populations of lymphocytes can be detected, one with the classic increased sensitivity to cross-linking agents characteristic of FA cells and the other usually a slowly increasing second population that is resistant to treatment with these agents. For a definition of genetic mosaicism see Box 7-7 . The presence of mosaicism is usually considered if the clinical course of the disease is milder than expected in comparison with individuals with the same genetic abnormality, if the course of the disease improves spontaneously rather than worsens, or in the presence of classic phenotypic abnormalities but rather mild or absent hematologic disease. The molecular mechanisms of genetic reversion include intragenic recombination (in compound heterozygotes), mitotic gene conversion (nonreciprocal exchange of genetic information as a result of heteroduplex formation between nonsister chromatids), DNA slippage, second-site compensating mutations, and site-specific correction by an as yet unknown mechanism. Gene reversion may occur in vivo or in vitro. In FA patients the reverting mutation is thought to occur in progenitor cells with self-renewing capability, possibly stem cells, thereby conferring a proliferative advantage to the revertant cells; this may lead to expansion of the revertant clone and progressive replacement of the FA cells in bone marrow. Genetic reversion in FA has been documented to date only in hematopoietic cells, not in fibroblasts. However, the fact that females heterozygous for FANCB mutations have biased X-inactivation in fibroblasts suggests that an intact FA pathway confers a proliferative advantage to nonhematopoietic cells. Thus the frequency of genetic reversion is largely unknown and so are its clinical consequences. In many patients genetic reversion is associated with hematologic improvement. However, genetic reversion has also been found in leukemic cells from FA patients, indicating that genetic reversion may confer a selective growth advantage and resistance of leukemic or preleukemic cells to specific chemotherapy drugs. Furthermore, genetic reversion in hematopoietic cells has no effect on cancer frequency in nonhematopoietic tissues. The strong selection for a functional FANC pathway may be used to advantage in gene therapy approaches. Genetic mosaicism is responsible at least in part for phenotypic variability, particularly within an individual family, and may create difficulty in interpretation of the chromosomal breakage testing with DEB and MMC (see the section on Diagnosis) or even lead to false-negative testing results. DEB or MMC testing of fibroblasts should therefore be considered in patients with clinical or genetic suspicion of FA but a negative DEB or MMC test result for peripheral blood lymphocytes.
Genetic mosaicism is defined as the presence of two or more genetically distinct populations of cells in a single individual that differ from each other at the DNA sequence level but are derived from a single zygote. In Fanconi anemia, genetic mosaicism results from reversion, which is the genetic correction of the FANC mutation.
The term attenuation has been introduced by Ceccaldi and associates for FA patients whose peripheral blood cells lack monoubiquitination of FANCD2 and have the characteristic increased chromosomal breaks after MMC treatment but whose cells, unlike those of most patients, do not show the characteristic G 2 arrest after MMC treatment. Most of these patients developed MDS or leukemia later in life. Attenuation was accompanied by clonal hematopoiesis, implying that all of the peripheral blood cells were derived from a single progenitor cell in which a molecular event took place that gave rise to attenuated G 2 arrest. The molecular mechanism underlying attenuation remains to be determined. Attenuation in patients with FA contributes to the variability in disease expression and identifies patients with a milder presentation of BMF but who might need continuous, intensified tumor surveillance.
Mouse Models for Fanconi Anemia
Orthologues of FANCD2, FANCM, and FANCL are found in nonvertebrates, whereas the other members of the FA core complex have evolved in vertebrates. To study clinical phenotypes and develop possible new therapeutic strategies, several mouse models for FA have been generated by introducing mutations into murine Fanc genes, including Fanca , Fancc , Fancg , Fancd2 , Fancd1, Fancf, Fancl, Fancm, Fancn, Fanco, and Fancp. Homozygous null mutations were lethal for Fancd1, Fancn, Fanco, and some Fancl embryonic mice, depending on the genetic background; this finding confirms what had already been suspected from the mutation analysis in patients with FA, namely that some residual function in these FANC genes is required for a living organism. Some models recapitulate the clinical features seen in patients with FA, specifically microphthalmos, germ cell defects, chromosomal instability, and increased sensitivity to cross-linking agents. In some models tumors develop with increased frequency, and tumor formation is more pronounced in the absence of p53. These models underline the importance of the FA pathway in tumorigenesis. Hematopoietic progenitor cells from mice homozygous for a null mutation in the Fancc gene demonstrate a distinct hypersensitivity to IFN-gamma that leads to cell death independent of the DNA repair pathway mediated by FAS-induced apoptosis, suggesting that IFN-gamma hypersensitivity may play a major role in the pathogenesis of BMF in patients with FA. Interestingly, however, although Fancd1- and Fancp- deficient mice have a reduced number of HSCs and decreased white blood cell and platelet counts, spontaneous development of aplastic anemia was not observed in these mice. Mice deficient in Fancd1 are exquisitely sensitive to excess acetaldehydes from endogenous and exogenous sources. Aldehydes are produced endogenously during normal metabolism or as a by-product of ethanol metabolism and can induce DNA-DNA and DNA-protein cross-links. Aldehydes are detoxified by aldehyde dehydrogenases. Interestingly, hematopoietic stem cells in mice express the aldehyde dehydrogenase Aldh2 , which is essential for protection against acetaldehyde toxicity, suggesting that the FA pathway is needed to protect from aldehyde-mediated genotoxicity. Although these findings need to be confirmed in humans, they would explain why hematopoiesis is preferentially affected in FA but is not affected, or not to the same extent, in other non-FA DNA repair disorders.
Induced Pluripotent Stem Cells Derived from Patients with Fanconi Anemia
Induced pluripotent stem cells (iPSCs) are derived from human somatic cells through ectopic expression of transcription factors. Since their discovery these cells have become a widely used tool for modeling and studying IBMFSs. Their unlimited self-renewal capacity, pluripotency, and ease of accessibility are very promising for disease modeling, cell-based therapy, and drug discovery. Interestingly, the generation of iPSCs from FA patients is challenging due to a low reprogramming efficiency in the presence of a defective FA pathway, suggesting that cellular reprogramming or the methodology (integrating viruses) of reprogramming requires an intact FA pathway. FA iPSC lines are capable of undergoing hematopoietic differentiation, but the hematopoietic progenitors undergo increased apoptosis and have a reduced clonogenic potential. Further investigation using the iPSC FA disease model is likely to provide exciting insights into the disease pathology and future treatment options.
Clinical Management
Once the diagnosis is confirmed, the family should be referred to a clinical geneticist for counseling and careful examination of family members. Genetic testing or chromosome breakage analysis should be offered to all family members. Early diagnosis is important for correct management of hematologic complications, diagnosis and appropriate treatment of coexisting congenital abnormalities and associated endocrinopathies, and identification of affected but asymptomatic family members and unaffected family members or pregnancies that may serve as potential hematopoietic stem cell donors. In addition, early diagnosis allows targeted cancer surveillance, not only in patients with FA but also in some cases in heterozygous mutation carriers (also see earlier). Management of patients in whom FA is diagnosed depends on the age at diagnosis and the presence or absence of congenital or hematologic abnormalities (see also Fanconi Anemia: Guidelines for Diagnosis and Management, Third Edition, 2008; available from the Fanconi Anemia Research Fund at fanconi.org/). All diagnosed FA patients should have a full hematologic assessment, including examination of bone marrow, and HLA typing should be performed in anticipation of possible HSCT. Other investigations at diagnosis should include audiometry, ophthalmic examination, renal ultrasound studies, and endocrine assessment. Referral to specialized surgeons might be necessary for correction of radial ray defects.
Management of Hematologic Disease
Initially, in the absence of hematologic abnormalities or clinical and laboratory signs of mild BMF, monitoring of peripheral blood values and yearly examination of bone marrow might be sufficient. Bone marrow examination should include a trephine biopsy and marrow aspirate for examination of marrow cellularity and morphologic changes characteristic of MDS, as well as for cytogenetic examination by G banding and fluorescent in situ hybridization (FISH) analysis, including probes that identify the most frequent cytogenetic abnormalities seen in patients with FA and progression to MDS or AML (see the section on Myelodysplastic Syndrome and Acute Leukemia in Patients with Fanconi Anemia). Plans for possible HSCT should be in place because hematologic complications may develop rapidly. With progression of BMF it will become necessary to increase the frequency of hematologic monitoring. HSCT is usually considered in patients with moderate BMF and an available HLA-matched unaffected sibling donor or when BMF becomes severe and an unrelated donor is available. Preventive HSCT in the absence of hematologic disease is highly controversial and currently considered investigational because the risk and time course of disease progression are highly variable and have to be weighed against the morbidity and mortality associated with HSCT. However preventive HSCT might be considered in patients with biallelic BRCA2 mutations associated with a higher risk of leukemic transformation.
Management of FA patients with MDS or AML, or both, is less standardized. The significance of transient clonal cytogenetic abnormalities limited to a single chromosome is unclear, but if they are persistent or complex, such abnormalities may predict a poor prognosis; the exception is an isolated trisomy 1q that might be present years before the development of MDS (see the section on Myelodysplastic Syndrome and Acute Leukemia in Patients with Fanconi Anemia). HSCT with or without induction chemotherapy or enrollment in clinical trials for MDS or MDS/AML is usually considered. Androgens such as oxymetholone, methyl testosterone, or danazol and hematopoietic growth factors transiently improve BMF in 50% to 60% of FA patients. Side effects of androgen therapy include masculinization, acne, hyperactivity, growth spurt followed by premature closure of the epiphyseal growth plates, high serum lipid levels, liver enzyme abnormalities, hepatic adenomas (with danazol), and a risk for hepatic adenocarcinomas (with androgens) that requires frequent monitoring of liver function and liver ultrasound scanning. The effect of pretransplantation androgen use is controversial. Hematopoietic growth factors [granulocyte colony-stimulating factor (G-CSF) or granulocyte-macrophage colony-stimulating factor (GM-CSF)] may transiently improve neutrophil counts and, in rare FA patients, hemoglobin levels and platelet numbers. The use of hematopoietic growth factors in patients with clonal cytogenetic abnormalities is controversial because of the potential risk of inducing or promoting leukemia. Antiinflammatory agents such as anti-TNF-alpha and antioxidants have been shown to improve hematopoiesis in vitro and in mouse models and are currently being tested in clinical trials for efficacy and safety in patients with FA. Red cell transfusion therapy and iron chelation (after multiple red cell transfusions) are frequently used in symptomatic FA patients. Platelet transfusions may be indicated in thrombocytopenic patients with bleeding or before surgical procedures.
HSCT from an HLA-matched sibling donor is accepted as the best treatment to cure BMF in FA patients and to prevent progression to MDS and AML. Early experiences with HSCT for the treatment of BMF were disastrous because of excessive regimen-related toxicity. Early conditioning regimens consisted of cyclophosphamide alone or in combination with total-body irradiation (TBI). Unfortunately, severe toxicities (e.g., gastrointestinal hemorrhage, hemorrhagic cystitis, cardiomyopathy, and skin burns) were common. Gluckman and colleagues proposed the use of a less intensive conditioning regimen consisting of low-dose cyclophosphamide and thoracoabdominal irradiation for patients with FA. This approach proved to be a major advance in the treatment of patients who had HLA-matched sibling donors. However, it did not afford consistent engraftment in recipients of unrelated donor transplants, with graft rejection rates exceeding 20%, and head and neck cancer was frequent after HSCT. Consequently, HSCT without irradiation is increasingly used for patients with FA and a matched sibling donor using fludarabine and cyclophosphamide and with T cell depletion either of the graft or in vivo with rabbit ATG or alemtuzumab to prevent graft-versus-host disease (GVHD). Recent studies suggest that fludarabine-based conditioning regimens are associated with improved survival rates in patients undergoing related or unrelated donor HSCT and have significantly improved survival rates for alternative donor transplant recipients. The use of irradiation for alternative donor transplant varies among countries and transplant centers. Transplant-related mortality and GVHD remain a problem. Furthermore chronic GVHD significantly increases the risk of squamous cell carcinoma in FA patients who undergo HSCT.
PGD may be used in parents with a previously affected child to select embryos that are both unaffected by FA (if a FANC gene mutation is known) and have HLA matched to the affected sibling. HSCT is performed with the HLA-matched umbilical cord blood of the unaffected newborn.
Unrelated cord blood as an alternative HSC source showed an overall survival rate of 53% with fludarabine in the conditioning regimen (vs. a rate of 19% without fludarabine). There was a relatively high incidence of GVHD and transplant-related mortality rates of 32% and 59%, respectively, suggesting that unrelated cord blood transplant should be considered only when other alternative donor stem cell sources cannot be found. Alternatively a mismatched or haploidentical family donor may be a valid option if no matched unrelated donor can be found.
Cancer Surveillance and Treatment
For FA patients who survive the hematologic complications, follow-up surveillance for solid malignant tumors becomes increasingly important. There are no clear guidelines on the treatment of cancer in FA patients, which is complicated because the increased sensitivity to irradiation and certain chemotherapeutic agents may result in enhanced toxicity and an increased risk of therapy-induced secondary malignant diseases.
Gene Therapy
HSCT may cure hematologic disease in FA patients; however, HLA-matched sibling donor cells are available only for a minority of patients. Gene therapy to correct HSCs, followed by autologous transplantation, offers an attractive alternative for the treatment of hematologic disease. Indeed, gene transfer corrects the hypersensitivity of most FA cell lines to DNA cross-linking agents in vitro (with the exception of FANCM, most likely because of the insolubility of the protein) and the hypersensitivity of FA knockout mice in vivo. The high incidence of somatic mosaicism and the often ameliorated course of hematologic disease in these patients (frequently referred to as the gene therapy of nature ) suggest that FA cells corrected by gene therapy are likely to have a selective growth and survival advantage. However, gene therapy attempts in FA patients have thus far been rather disappointing, mainly because of the lack of permanent gene correction and the difficulty in culturing FA bone marrow cells in vitro. Multiple investigations are currently addressing the major difficulties of gene therapy for FA, including the scarcity of hematopoietic stem cells in patients with BMF, the extraordinary fragility of FA hematopoietic progenitor cells, and the increased sensitivity of FA hematopoietic progenitor cells to oxidative stress, which makes it difficult to culture and transduce FA hematopoietic progenitor cells in vitro. There is also a risk that the transduced hematopoietic cells might already have acquired secondary somatic, and possibly oncogenic, mutations and that correction by gene therapy would promote rather than prevent leukemogenesis. The International FA Gene Therapy Working Group was formed to accelerate the transition from research to clinical gene therapy trials and coordinating activities on an international scale.
Dyskeratosis Congenita
Dyskeratosis congenita (DC), also known as Zinsser-Cole-Engman syndrome , is genetically and phenotypically heterogeneous ( Box 7-8 ). It was first described by Zinsser and recognized as a clinical entity by Cole and Engman. DC was originally perceived to be a dermatologic disorder affecting the skin, nails, and mouth. Its association with BMF and squamous cell cancer arising from the skin or from leukoplakia in the mouth or anus was recognized later.
Dyskeratosis congenita is a rare inherited bone marrow failure syndrome with an X-linked, autosomal dominant, and autosomal recessive inheritance. However, more than half of patients present with sporadic disease. Classically, bone marrow failure in these dyskeratosis congenita patients is associated with the mucocutaneous triad including abnormal pigmentation, dystrophic nails, and mucosal leukoplakia. Patients with dyskeratosis congenita have a defect in telomere maintenance, and at the time of bone marrow failure peripheral leukocytes have very short telomeres (see Fig. 7-9 ).
Clinical Manifestations
Originally, DC was clinically recognized by the classic diagnostic triad of reticular pigmentation of the skin, nail dystrophy, and mucosal leukoplakia ( Figure 7-7 ). Other signs and symptoms found frequently in classic DC are summarized in Table 7-6 . The classic clinical course is mucocutaneous features first appearing in childhood, BMF developing in adolescence, and death occurring in young adulthood. Cytopenias due to BMF, immune deficiency, pulmonary fibrosis, and malignant disease are the major causes of death.
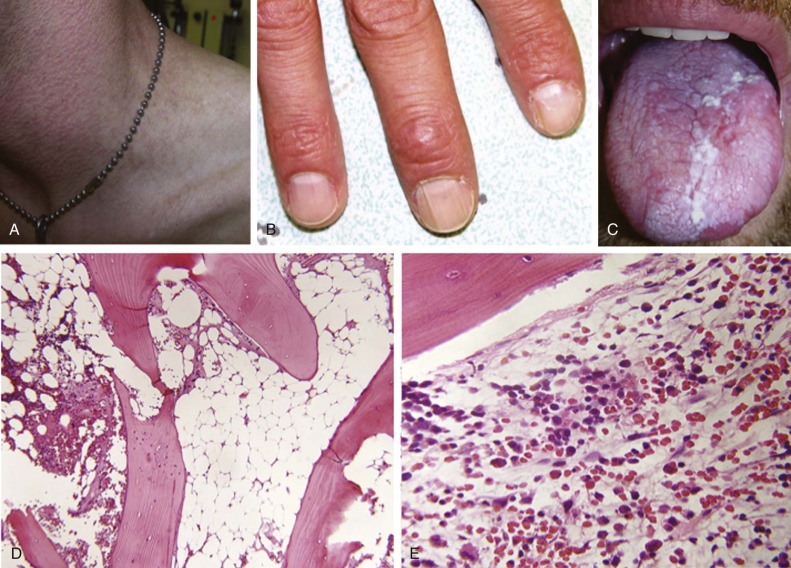
| Characteristic | Alter 2003 (%) | Vullamy & Dokal 2006 (%) |
|---|---|---|
| Diagnostic mucocutaneous features | ||
| Skin pigmentation | 88 | 89 |
| Nail dystrophy | 73 | 88 |
| Leukoplakia | 64 | 78 |
| Eye abnormalities/epiphora | 38 | 31 |
| Teeth abnormalities/caries/loss | 19 | 17 |
| Learning difficulties/developmental delay/cognitive disability | 14 | 25 |
| Pulmonary diease/fibrosis | 20 | |
| Skeletal anomalies | 14 | |
| osteoporosis/avascular necrosis/scoliosis | 5 | |
| Short stature | 14 | 20 |
| Hyperhidrosis | 10 | 15 |
| Hair loss, early graying | 18 | 16 |
| Urinary tract abnormalities | 7 | |
| Urethral stricture/phimosis | 5 | |
| Gastrointestinal abnormalities | 14 | |
| Liver fibrosis/ulcer/enteropathy | 7 | |
| Esophageal stricture | 17 | |
| Other conditions | 11 | |
| Gonadal anomalies/hypogonadism/undescended testes | 5 | 6 |
| Intrauterine growth retardation | ||
| Ataxia/cerebellar hypoplasia | 7 | |
| Microencephaly | 8 | |
| Malignant disease | 12 | 10 |
The identification of nine of the genes responsible for DC and the realization that these genes converge into a common pathway have changed the perception of this disease. It is now thought that individuals in whom DC is diagnosed on the basis of BMF and the classic cutaneous manifestations represent only a fraction of those who suffer from this condition. Whether affected individuals in the absence of cutaneous disease should be labeled with the diagnosis of “dyskeratosis congenita” or whether the disease should be renamed with a term better reflecting the pathogenetic pathway is still under debate. Here we will use the conventional nomenclature and clinically classify patients as having classic, atypical, or silent DC. Patients who have two or more of the three diagnostic mucocutaneous features or who have one of the mucocutaneous features and hypocellular bone marrow with all three blood cell lineages affected are those with classic DC. Patients with BMF but lacking mucocutaneous features or patients with clinical manifestations of DC other than hematologic disease or mucocutaneous features, such as pulmonary fibrosis, are those with atypical DC. Individuals with no clinical manifestations but carrying a pathogenic mutation are classified as having silent DC. Our knowledge about the natural history of DC is biased because early studies focused on patients with the classic clinical manifestations. Our experience is still limited about the course of disease, response to treatment, and late complications in individuals with atypical or silent DC.
Classic Dyskeratosis Congenita
Mucocutaneous Features.
Mucocutaneous abnormalities are the hallmark of classic DC (see Fig. 7-7 ). The most commonly observed mucocutaneous abnormality (>90%) is reticulated skin hyperpigmentation on the neck, face, chest, and arms. It may be extensive or localized and tends to increase with age. Other cutaneous manifestations include alopecia, sparse hair, premature hair graying, hyperhidrosis, hyperkeratosis of the palms and soles, and loss of dermal ridges on the fingers and toes.
Nail dystrophy is another frequent finding (>80%). Progressive nail dystrophy starts with ridging and longitudinal splitting and may proceed via atrophy and thinning to complete nail loss. The severity of nail dystrophy may vary among the digits; fingernails are usually more severely affected than toenails.
Mucosal leukoplakia occurs in the majority of patients (>70%) and typically involves the tongue, buccal mucosa, and oropharynx, with the tongue being the most frequently affected site. Mucosal leukoplakia may be the initial clinical manifestation of disease. Consequently, dentists or oral surgeons may be the first health professionals consulted. Histologic examination reveals hyperkeratosis or a lichenoid reaction. Other sites of leukoplakia include the urethra, glans penis, vagina, and anorectal region. DC patients have an increased risk of malignant disease arising from the preexisting leukoplakia; the incidence of malignant transformation has been estimated to be 35%.
Hematologic Abnormalities.
About 85% of patients with classic DC are initially found to have cytopenia of one or more lineages, and pancytopenia develops in more than 95% by 40 years of age. The overall cumulative incidence of BMF in all patients with DC (typical and atypical) is approximately 50%. Often the degree of aplasia in the bone marrow is striking despite only a moderate degree of peripheral blood cytopenia. Complications of BMF, such as hemorrhage or opportunistic infection, represent the major causes of death in patients with DC. In patients with atypical DC, BMF may appear before the classic mucocutaneous features and lead to an initial diagnosis of idiopathic aplastic anemia. Hypoplastic MDS and MDS/AML are increasingly diagnosed in patients with DC, especially in those with a milder course or an atypical form of the disease. Indeed, MDS or MDS/AML may be the initial manifestation of patients with atypical DC.
Combined immune deficiency is frequently associated with DC, particularly in children with severe and early onset of disease. Reduced or absent B cells and reduced NK cells are frequent; other findings in young patients include decreased T-cell numbers, low immunoglobulin levels, and a lack of lymphocyte proliferation in response to antigens.
Other Clinical Features.
Pulmonary complications are common (>20%) and include pulmonary fibrosis and vascular abnormalities. Pulmonary fibrosis is usually a more prominent finding in older patients or after BMT. In some patients with atypical DC, pulmonary fibrosis may be the only clinical manifestation. Figure 7-8 shows a family with autosomal dominant DC and pulmonary fibrosis. Families with hereditary pulmonary fibrosis of adult onset should be investigated for DC, particularly when pulmonary fibrosis is associated with macrocytic anemia or other cytopenias. In families with pulmonary fibrosis and BMF, a pathogenic DC gene mutation is almost always responsible for both conditions; in such kindreds pulmonary disease typically manifests in the sixth decade of life, whereas BMF manifests in the first or second decade (see Fig. 7-8 ).
Liver fibrosis may be a late manifestation of DC. As with pulmonary fibrosis, liver fibrosis tends to occur in older patients or after BMT. In some patients liver fibrosis might be the only or first manifestation of disease. Excessive alcohol consumption or acute hepatitis is thought to accelerate liver fibrosis and severity in patients with a pathogenic DC mutation. The presence of a DC mutation does not preclude the development of hepatocellular carcinoma.
Ocular findings include epiphora (excessive tearing caused by blocked lacrimal ducts), blepharitis, cataracts, loss of eyelashes, conjunctivitis, ectropion, glaucoma, strabismus, ulcers, ocular albinism, and exudative retinopathy ( Coats retinopathy ). Interestingly, Coats retinopathy is specific for Revesz syndrome (caused by mutations in TINF2 ) and Coats- plus syndrome (caused by mutations in CTC1 ); exudative retinopathy is not typical of other forms of DC. Corneal limbal insufficiency owing to impaired corneal stem cell function has also been described in patients with DC.
Sparse hair and early graying are common. Dilated cardiomyopathy due to cardiac fibrosis has been associated with DC.
Multiple dental caries early loss of teeth, and a bifid uvula are common oral manifestations of DC. Gastrointestinal abnormalities include web formation in the upper esophagus, esophageal stricture, severe necrotizing ulcerative pancolitis in infants, and chronic diarrhea with malabsorption due to mucosal villous blunting in young adults. Patients have an increased risk of esophageal, rectal, and possibly gastric cancers.
Genitourinary abnormalities include hypospadias, phimosis, and urethral strictures. Underdeveloped testes and azoospermia have been reported in more severe and advanced forms of DC.
Cognitive deficiencies such as learning disorders are common in classic DC. In rare cases an association with schizophrenia has been described. Other neurologic manifestations may include cerebellar ataxia due to cerebellar hypoplasia, intracranial calcification, and hypoplasia of the corpus callosum, pituitary gland, or brainstem.
Osteoporosis and avascular necrosis of the bones, in particular the hips, is frequent and probably underappreciated and may complicate the course of the disease. In some forms of DC avascular necrosis of the bones occurs at a very young age and may be the dominating clinical feature. Short stature and an elf-like appearance have been associated with classic DC.
Predisposition to Malignant Disease
DC is a syndrome that predisposes to cancer. After BMF and pulmonary complications, malignant disease is the third leading cause of death. The cumulative incidence has been estimated to be 20% to 30% by the age of 50 years. The most frequent malignant tumors in patients with DC are squamous cell carcinoma of the head and neck (especially the tongue but also the lip, mouth, palate, cheek, nasopharynx, and larynx), the upper and lower gastrointestinal tract (esophagus and anus), and the female genital tract. Other reported malignant tumors include skin cancer and adenocarcinoma of the stomach, colon, lung, and pancreas. Whether all forms of DC have equal risks of predisposition to cancer remains to be determined. Malignant tumors usually develop in the third decade and are therefore more frequently found in individuals with milder and late-onset forms of the disease (see Table 7-1 ).
MDS and MDS/AML are common in patients with atypical DC and in patients with late-onset of the disease (see Fig. 7-7, E ). The cumulative incidence of MDS has been estimated to be about 30% and that of MDS/AML approximately 10% by the age of 50 years. MDS or MDS/AML may be the first hematologic manifestation of disease. Although DC is rarely a cause of MDS or MDS/AML in children, it should be considered in the differential diagnosis in young adults with MDS or MDS/AML. Abnormalities of chromosome 7 (monosomy, isochromosome, or other structural rearrangements) are the most common cytogenetic abnormalities in patients with DC-associated MDS or MDS/AML.
Little is known about the biology of leukemia or other cancers in patients with DC and to what extent these malignant diseases differ from those seen in patients without DC. The cancer recurrence rate is high in patients with DC.
Laboratory Findings
Common laboratory findings are summarized in Box 7-9 . In most patients, the initial hematologic abnormalities are thrombocytopenia or macrocytic anemia followed by pancytopenia. Bone marrow examination often reveals increased cellularity at the outset, suggesting an element of hypersplenism. However, hypocellularity then ensues, consistent with the diagnosis of aplastic anemia. Macrocytosis and elevated hemoglobin F levels (stress erythropoiesis) are common. The number of circulating hematopoietic progenitor cells is decreased, indicative of the impairment in hematopoiesis at the stem cell level.
* Laboratory findings in bold are characteristic for dyskeratosis congenita.
- 1.
Peripheral blood findings
Cytopenia of one or more lineages (80%):
Initial presentation highly variable:
macrocytosis +/− anemia
Thrombocytopenia
Neutropenia
Pancytopenia
Low number of circulating progenitor cells
Elevated hemoglobin F
Elevated von Willebrand factor
- 2.
Bone marrow examination
Hypocellular bone marrow affecting all three lineages
Increased number of mast cells
Dyserythropoiesis
Hypocellular myelodysplastic syndrome
Myelodysplastic syndrome/acute myeloid leukemia
- 3.
Immunologic findings
Humoral abnormalities:
Hypogammaglobulinemia or hypergammaglobulinemia
Cellular abnormalities:
Lymphopenia: B cells, T cells, NK cells
Functional abnormalities:
Proliferation upon stimulation is reduced
Increased apoptosis upon stimulation
Anergy
Difficulty in immortalization by Epstein-Barr virus transformation
- 4.
Cellular abnormalities
Premature replicative senescence
No increased chromosomal breakage after cross-linking agents
Hypersensitivity to ionizing irradiation (controversial)
Increased sensitivity to certain cytotoxic agents
Difficulty in immortalization by TERT
Difficulty in reprogramming to induce pluripotent stem cells
- 5.
Cytogenetic abnormalities
Chromosomal instability: Nonclonal complex anomalies in cultured cells:
Chromosomal breaks, hypodiploidy, micronuclei,
anaphase and telophase bridges,
short telomeres/signal free ends
end-to-end fusion with no telomeric signal
No increase in chromsomal instability after clastogenic effects of DNA
cross-linking agents
Biased X-inactivation (female carrier of X-linked dyskeratosis congenita)
Clonal abnormalities in bone marrow cells : monosomy 7 (myelodysplastic syndrome, myelodysplastic syndrome/acute lymphocytic leukemia)
Bone marrow examination usually shows hypocellular marrow affecting all three blood cell lineages. The hypocellularity of the bone marrow is often striking in comparison with rather moderate cytopenias in the peripheral blood. Hypocellular MDS or MDS/AML is increasingly being found in patients with atypical DC or a late onset of disease. Monosomy 7 is the most frequent clonal cytogenetic abnormality in patients with MDS or MDS/AML.
Long-term culture assays of bone marrow cells show a decreased number of progenitor cells and an impaired capability for colony formation, indicative of a quantitative and qualitative defect.
Immunologic abnormalities may include humoral and cellular defects (summarized in Box 7-9 ). Severe combined immunodeficiency is characteristic of severe forms of DC.
DC cells have increased spontaneous chromosomal instability that leads to distinctive chromosomal breaks, rearrangements, and sister chromatid exchanges. Chromosomal instability increases as DC cells are passaged in vitro. Chromosome breakage does not increase after treatment with alkylating agents such as DEB, MMC, or nitrogen mustard, a finding that distinguishes DC from FA. Chromosomal abnormalities characteristic of DC include end-to-end fusions, dicentric or tricentric chromosomes, anaphase or telophase bridges, short telomeres, and telomere free ends.
Paternally or maternally biased X-inactivation in blood cells, as well as in fibroblasts from buccal mucosa and skin biopsy samples in female mutation carriers, is characteristic of X-linked DC.
The presence of short telomeres in circulating peripheral blood cells is a hallmark of patients with BMF and DC. Telomere length in individuals with BMF secondary to DC is far below the normal distribution of telomere length in age-matched healthy control individuals ( Box 7-10 and Fig. 7-9 ). In contrast to other IBMFSs, which may have short telomeres in circulating myeloid cells, telomeres are exquisitely short in the myeloid and lymphoid cells of patients with DC-associated BMF. Telomere length measurements of circulating peripheral blood lymphocytes (or total blood cells, although less sensitive) are therefore used to screen patients with BMF for DC; normal telomere length makes DC an unlikely cause of BMF. Similarly, very short telomeres in circulating lymphocytes are highly suggestive of DC, but the diagnosis should be reserved for patients with classic clinical DC features or an identified pathogenic DC mutation.
Telomeres are complex DNA/protein structures at the ends of chromosomes that are needed to protect chromosome ends from degradation and distinguish them from double-stranded breaks. Human telomere DNA consists of thousands of repeats of TTAGGG (see Fig. 7-10, A and B ). Most somatic cells lack telomerase, so telomeres shorten with each cell division owing to the inability of DNA polymerase I to copy the extreme end of a DNA strand. In most tissues, therefore, telomere length decreases with age. When telomeres become critically short, cell cycle arrest or cell death occurs (see Fig. 7-11 ). In germ cells , some stem cells , and most cancer cells , telomeres are maintained by the action of the enzyme telomerase , which contains two core components, a reverse transcriptase TERT (telomerase reverse transcriptase) and an RNA molecule, TERC (telomerase RNA component), which acts as a template for the synthesis of telomere repeats (see Fig. 7-10, C and D ).
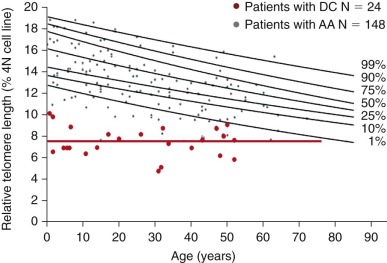
Cellular Phenotype of Dyskeratosis Congenita Cells
In addition to increased spontaneous chromosomal instability, DC cells exhibit several other phenotypic characteristics, as summarized in Box 7-9 . DC cells have been reported to have increased sensitivity to ionizing radiation and certain chemotherapeutic agents (e.g., bleomycin), although these findings were not consistent among all investigators. Decreased proliferation capacity in vitro and premature replicative senescence are also characteristic of DC cells (see Box 7-9 ). Cells from classic and more severe forms of DC (X-linked DC) fail to immortalize after Epstein-Barr virus–induced transformation or after transfection of a plasmid expressing the telomerase catalytic subunit TERT. Similarly the reprogramming of DC skin cells into iPS cells is difficult, and reprogrammed iPS cells are not immortal.
Differential Diagnosis
Clinically, BMF in patients with DC has to be distinguished from other forms of BMF, including acquired aplastic anemia, familial aplastic anemia, and IBMFSs such as FA (MIM 227650), Seckel syndrome (MIM 606593), DNA ligase IV deficiency (MIM 606593), and Shwachman-Diamond syndrome (SDS, MIM 260400). Reticulate hyperpigmented disorders including Naegeli-Franceschetti-Jadassohn syndrome (MIM 161000), dermatopathia pigmentosa reticularis (MIM 125595), X-linked reticulate pigmentary disorder, incontinentia pigmenti (MIM 308300), and poikiloderma with neutropenia (MIM 604173) need to be distinguished from DC. Clinical manifestations, associated physical abnormalities, the family history (pattern of inheritance), and the lack of increased chromosomal fragility in the DEB or MMC test performed on cultured peripheral lymphocytes or fibroblasts (or both) are helpful in strengthening the diagnosis of DC. As noted earlier, telomere length below the normal distribution strongly suggests DC, whereas normal telomere length makes DC an unlikely cause for BMF in these patients. X-inactivation studies may be helpful in identifying female carriers of the X-linked form of DC.
Genetic Characteristics
Inheritance.
There are four distinct forms of inheritance of DC. Historically, before molecular diagnostic testing was available, inheritance was determined by examining family members for the presence of clinical manifestations. Today, we know that such classification was often mistaken because of the variability of penetrance of gene mutations.
X-linked Dyskeratosis Congenita.
The most common form of classic DC is the X-linked form (MIM 30500), and many families with classic DC and X-linked inheritance have been described. The X-linked form is almost always due to single point mutations in the DKC1 gene encoding dyskerin.
Autosomal Dominant Dyskeratosis Congenita.
Several families have been described with an autosomal dominant (AD) form of inheritance of DC (AD DC, MIM 127550). This form exhibits genetic anticipation ( Box 7-11 ). Several pedigrees in which AD DC is caused by mutations in the TERC gene encoding telomerase RNA have been described. In addition, families in whom AD DC appears to be due to mutations in TERT , which encodes the telomerase reverse transcriptase, have been reported. Mutations in the shelterin component TIN2, encoded by TINF2 , are dominant in that a severe form of DC is caused by heterozygous mutations. However, these patients seldom have children, and the disease is sporadic, caused by de novo mutations.
Autosomal Recessive Dyskeratosis Congenita.
A number of families have been described with autosomal recessive (AR) DC (MIM 224230). AR DC is usually inferred when neither parent has any signs of DC but two or more children have the disease or when the parents of an affected child are consanguineous. Some families originally thought to have AR DC were found to have AD DC with variable penetrance and expressivity, mimicking sporadic or AR disease. Rare cases of AR DC are due to biallelic mutations in TERT, TERC, NOP10, NHP2 , and WRAP53 . More commonly, recessive forms of DC are due to mutations in CTC1 and RTEL1 .
Sporadic Dyskeratosis Congenita.
A large proportion of DC cases arise sporadically (i.e., neither parent of an affected child has the disease). Of 123 such male cases from the DC registry in the United Kingdom, 40 were found to have a de novo DKC1 mutation and 3 were found to have an inherited TERC mutation with variable penetrance and expressivity in the affected family. Eighty had neither TERC nor DKC1 mutations. Similarly, all 35 female cases had neither TERC nor DKC1 mutations. These sporadic cases account for more than half of DC patients. Most cases due to TINF2 mutations are sporadic, due to the fact that most patients with this severe form of DC do not reproduce.
Dyskeratosis Congenita Genes and Their Functions
DKC1 and Dyskerin.
X-linked DC is caused by mutations in the DKC1 gene at Xq28. DKC1 was identified by positional cloning in families with affected males and biased X-inactivation in female carriers. The DKC1 -encoded protein dyskerin is a 58-kD nucleolar protein orthologous to proteins in yeast, flies, and rats (Cbf5p, Nop60B/minifly, and NAP57, respectively).
Dyskerin is associated with box H/ACA RNA molecules in ribonucleoprotein (RNP) complexes. Box H/ACA RNP complexes consist of four different proteins (dyskerin, GAR1, NOP10, and NHP2) and an integral RNA species. The box H/ACA RNA determines the function of the RNP complex. The majority of box H/ACA RNAs are small nucleolar RNAs (snoRNAs) found in the nucleolus. These snoRNAs guide the protein complex containing dyskerin by complementary base pairing to specific uridine residues of newly synthesized ribosomal RNA (rRNA) molecules, which are then modified by pseudouridylation. In snoRNP dyskerin is the enzyme, a pseudouridine synthase, that actually catalyzes the pseudouridylation reaction. The role of pseudouridylation is not known, but pseudouridines tend to be found in the conserved sequences of rRNA and transfer RNA (tRNA) in regions of high secondary structure, and it is likely that these modifications stabilize the secondary and tertiary structures of RNA molecules. Other box H/ACA RNAs that associate with dyskerin and the three other proteins of the complex participate in ribosome biogenesis downstream of rRNA modification or messenger RNA (mRNA) splicing or are orphan RNAs, the function of which remains to be determined.
Telomerase RNA ( TERC , telomerase RNA component), which along with TERT (telomerase reverse transcriptase) forms the core of the telomerase enzyme, also associates with dyskerin and the other three proteins present in H/ACA-class snoRNP ( Fig. 7-10, C ). The conserved secondary structure of the 3′ end of telomerase RNA is very similar to that of H/ACA RNAs. This part of the telomerase RNA and its association with H/ACA RNP components are found only in vertebrates, whereas the H/ACA RNP components themselves are highly conserved among all eukaryotes. The four proteins may be involved in transport, stability, and assembly of the telomerase complex in the nucleolus. There is no evidence that the H/ACA part of telomerase RNA functions as a guide RNA or that dyskerin exhibits any pseudouridylation activity as part of the telomerase complex.
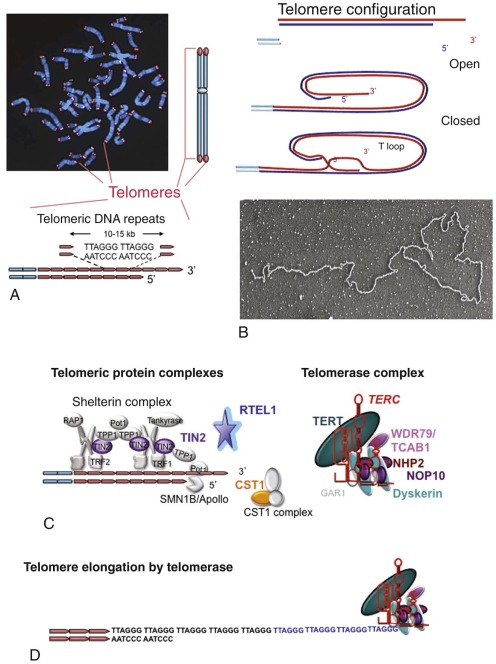
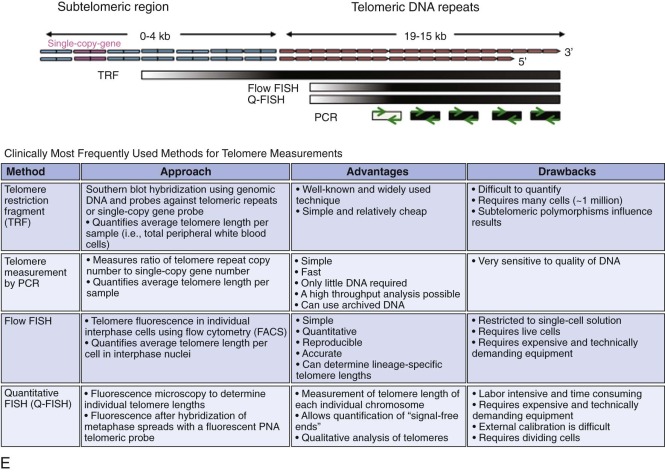
The diverse function of dyskerin in humans raises the interesting question of whether and to what extent the pathogenesis of X-linked DC is caused by defects in telomerase activity or in other functions of RNP, such as rRNA metabolism.
NOP10.
Autozygous mutations (homozygous mutations in a consanguineous family) in the NOP10 gene on chromosome 15q14 have been found to be responsible for disease in a single family. NOP10, like dyskerin, is an essential component of H/ACA RNP complexes and thus participates in the same pathways as dyskerin (see Fig. 7-10, C ).
NHP2.
Two families have been described with biallelic mutations in NHP2 . NHP2, like dyskerin and NOP10, is an essential component of H/ACA snoRNP complexes (see Fig. 7-10, C ).
TERC, TERT, and Telomerase.
AD DC is caused by mutations in the TERC gene, also abbreviated as hTR or TER , which is found on chromosome 3q26. TERC was identified as the gene responsible for AD DC through positional cloning. It encodes the RNA component of the telomerase complex. Telomerase, the enzyme responsible for elongating and maintaining telomeres, synthesizes telomeric DNA repeats by using TERC RNA as a template (see Box 7-10 and Fig. 7-10, C and D ). Nonsynonymous mutations have also been identified in the TERT gene localized on 5p15.33. TERT is the catalytic part of the telomerase complex (see Box 7-10 and Fig. 7-10, C ). TERT was identified as a gene responsible for AD DC through a candidate gene approach. Telomerase RNA, or the TERC component, which along with TERT forms the core of the telomerase enzyme, also associates with the four proteins present in H/ACA-class RNPs, including dyskerin, NOP10, and NHP2 (telomerase RNP complex; see Fig. 7-10, C ). Dyskerin, TERT, and TERC were the three components identified in purified active telomerase.
The measurement of telomere length has become a widely used screening test to identify patients who have BMF due to DC. Several methods are available that provide this type of information (for review, see Aubert et al, 2012, and Vera and Blasco, 2012 ). Although the methods in general produce similar results, individual measurements are difficult to compare. Methods most frequently used clinically include the measurement of telomere restriction fragment (TRF) lengths; PCR amplification of telomere repeats; and FISH, quantitative FISH (Q-FISH), and flow FISH, which are based on digital microscopy and flow cytometry, respectively (see Fig. 7-10, E ).
TINF2.
Heterozygous mutations in TINF2 represent the second most frequent cause of DC, after DKC1 mutations. TINF2 encodes TIN2, one of the six components of the shelterin complex, which interacts with telomere DNA repeats and protects the ends of chromosomes from degradation and from being recognized by the cell’s DNA repair machinery (see Fig. 7-10, C ). TIN2, along with another shelterin component, TPP1, is important in recruiting telomerase to its site of action at the telomere. TIN2 also associates with a protein called HP1-gamma, which binds to telomeres in S phase and is important in cohesion, the physical pairing of sister chromatids by association with proteins called cohesions. The HP1-gamma binding site in TIN2 is specifically required for cohesion of sister telomeres, and mutations in this site can affect telomere length maintenance.
TCAB1.
Two unrelated cases have been reported where DC was caused by compound heterozygous mutations in TCAB1. TCAB1, also known as WDR79 and WRAP53, contains WD40 repeats and binds to the CAB box, a motif found in scaRNAs and TERC (see Figs. 7-10, C and 7-12, C ) . Binding of TCAB1 is necessary for localization in Cajal bodies, the nuclear sites of assembly of telomerase and scaRNAs. Telomerase assembly in Cajal bodies is important in the translocation of telomerase to its site of action at the telomere, and TCAB1 inhibition leads to telomere shortening.
CTC1.
Patients with biallelic mutations in CTC1 (conserved telomere component 1) present with a variable set of abnormalities, including some characteristic features of DC. CTC1 forms a complex with two other proteins, Suppressor of cdc ThirteeN 1 (STN1) and TElomeric pathways with stN1 (TEN1). This complex is known as the CST telomere-capping complex (see Fig. 7-10, C ). CTC1 and STN1 stimulate DNA polymerase primase by binding single-stranded DNA, suggesting that the complex may be involved in priming telomeric C-strand synthesis. The corresponding complex in yeast is involved in maintaining telomeric DNA integrity. CTC1 may have a general role in DNA metabolism rather than a specifically telomeric one.
RTEL1.
Both homozygous and heterozygous mutations in RTEL1 have been described in DC patients. RTEL1 is a helicase originally identified in a screen of genes that affect telomere length. RTEL1 plays an essential role in maintaining telomere length and genome stability and is an antagonist of homologous recombination. It has recently been shown that RTEL1 is essential for the disassembly of telomeric T loops; without proper disassembly telomere loss occurs because of the action of the SLX4 nuclease complex, which resolves the T loop as a circle. G-quadruplex structures at the telomere are a major source of telomere fragility in RTEL1- deficient cells.
Other Genes.
For about 50% of patients in whom DC and BMF are diagnosed, the mutations or the genes mutated have not been identified. Like patients with DC and BMF caused by mutations in known genes, most of these patients, but not all, also have very short telomeres, suggesting that the affected gene or genes participate in the same pathway.
Unraveling of the molecular pathology of DC has greatly advanced our understanding of the disease. The finding that all forms of DC in which the pathogenesis is understood are caused by disruption of telomere maintenance highlights the importance of telomeres in the disease (see Fig. 7-9 ). Figure 7-11 summarizes our current model of the pathogenesis of DC.
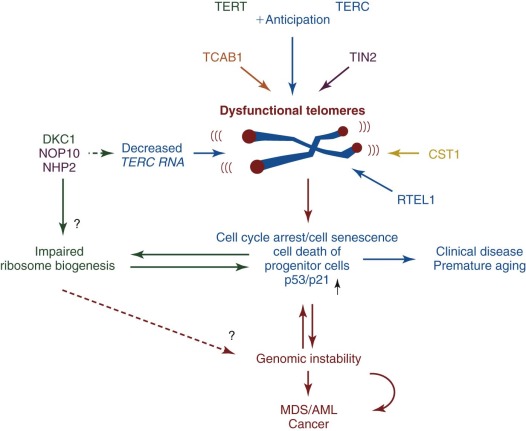
Dyskeratosis Congenita Gene Mutations
Mutations in telomere maintenance genes that have been found in patients with DC are listed on the comprehensive Telomerase Database, which is updated regularly.
DKC1 Mutations.
The mutations in the DKC1 gene that cause DC are mainly single–amino acid missense mutations ( Fig. 7-12, A ) and can be inherited or occur de novo. The A353V mutation is the most frequent de novo DKC1 mutation and is found in approximately 40% of patients with DC and DKC1 mutations. DKC1 mutations are not randomly distributed but are concentrated in the N-terminal region of dyskerin. The crystal structure of the archaeal homologue of the H/ACA RNP complex, the Cbf5-Nop10 complex, has enabled modeling of human dyskerin. In this model most of the residues mutated in cases of DC are tightly clustered in or near the PUA domain, which is predicted to bind H/ACA RNA, including TERC RNA (see Fig. 7-12, B ). The mutations would thus be expected to decrease binding of the cognate RNA and lead to instability. Only two mutations have been found in the TruB catalytic domain, one of which, S121G, is located three residues from the essential aspartate required for pseudouridylation and, interestingly, is associated with a severe form of DC known as Hoyeraal-Hreidarsson syndrome (HHS, MIM 300240). The absence of mutations that would predict a “null” allele, such as frameshifts and nonsense mutations, suggests that a dyskerin null mutation is incompatible with life. In support of this notion, a dyskerin null mutation is embryonically lethal in mice.
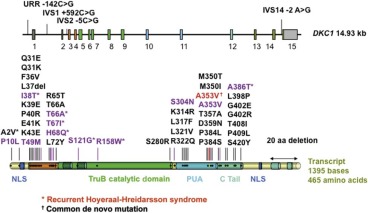
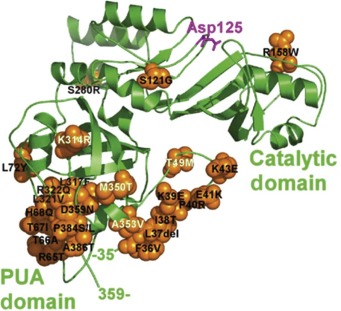
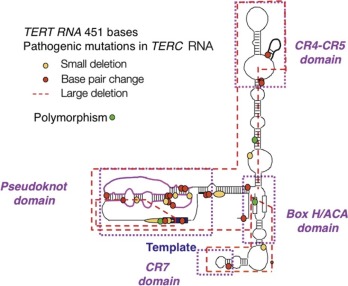
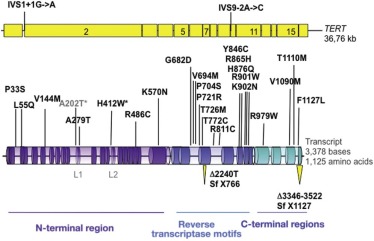
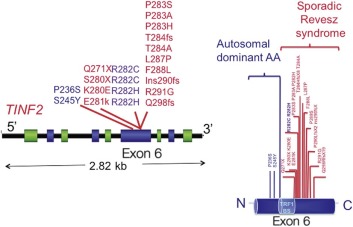
The functional consequences of a subset of pathogenic DKC1 mutations have been investigated in fibroblasts or mononuclear cells from patients with X-linked DC and in XY mouse embryonic stem cells harboring a mutated Dkc1 allele. Analysis of fibroblasts or mononuclear cells from patients with X-linked DC showed a reduced level of TERC RNA and decreased telomerase activity, whereas analysis in mouse cells revealed that not only the level of Terc RNA but also the levels of other specific snoRNAs were affected and that overall pseudouridylation activity—in particular, site-specific pseudouridylation involving the affected guide snoRNA—was markedly decreased. Controversy continues as to whether X-linked DC is due solely to defective telomere maintenance or whether defective ribosome biogenesis contributes to the pathogenesis. If the latter is the case, other relevant genes may not be effectively translated and thereby contribute to the pathophysiology (see later discussion of Diamond-Blackfan Anemia [DBA]).
Biochemical and structural studies of the assembly and structure of H/ACA RNPs, including telomerase, have so far failed to explain the structural and functional relationships in the effects of the DKC1 mutations. Early in the biogenesis of H/ACA RNPs, prior to introduction of NAF1 and before assembly with the RNA component, dyskerin forms a tight connection with a protein SHQ1 and many DKC1 mutations are in the region of dyskerin that interacts with SHQ1, suggesting that the mutations may affect biogenesis of the complex. It will be interesting to see if telomerase is preferentially affected by these mutations.
NOP10 Mutations.
Autozygous NOP10 mutations have been identified in one consanguineous family. The mutation leads to replacement of a highly conserved arginine with tryptophan at amino acid position 34 (R34W). The three affected family members were autozygous for the mutation. Heterozygous mutation carriers did not show obvious signs of disease but had shortened telomeres and decreased levels of TERC RNA in their peripheral blood cells. The inheritance of hypomorphic gene mutations may mimic a recessive inheritance pattern. The shortened telomeres and reduced levels of TERC RNA in heterozygous mutation carriers, however, suggest that both NOP10 alleles are expressed codominantly. Whether the mutation affects the function of only telomerase RNP or also other H/ACA RNP complexes remains to be determined. In vitro experiments showed that the mutated NOP10 protein was not defective in the formation of the dyskerin-NAF1-NOP10-NHP2 tetramer that assembles with the nascent H/ACA RNA; rather, the mutation severely affected association with the H/ACA domain of TERC and a subset of other H/ACA RNAs.
NHP2 Mutations.
Two families with NHP2 mutations have been reported. In one kindred affected children were homozygous for a Y139H mutation, while in the other the affected child was a compound heterozygote for a V126 M and a mutation in the stop codon, X154RextX*52, resulting in an elongated protein. In both cases parents were heterozygous and asymptomatic. Patients had low TERC levels and short telomeres. In vitro experiments showed that the V126M and the Y139H mutations impaired association with NOP10 and led to defects in H/ACA RNP assembly with all H/ACA RNAs tested, including TERC.
TERC Mutations.
The TERC mutations found in patients with DC are shown in Figure 7-12, C . Investigation of family members revealed that in all patients with clinical manifestations of DC the TERC gene mutations were inherited. Many mutations are found in the pseudoknot domain of the molecule, a domain that mediates binding to the telomerase reverse transcriptase TERT and is essential for telomerase activity. Other mutations are found in the CR7 and H/ACA domains and affect the stability and nuclear accumulation of the TERC molecule. In many of these mutations it has been demonstrated that the mutations act by disrupting the secondary structure of the TERC molecule rather than by altering nucleotide sequences crucial for function ; this finding is in agreement with the fact that although the secondary structure of TERC is highly conserved among different species, the primary sequence is not. Large gene deletions encompassing the TERC gene have been identified in two independent families with AD DC. Along with the pathogenic mutations, which have been shown by several groups to severely reduce telomerase activity, there are two polymorphisms that are relatively frequent in people of African descent. These sequence changes, G58A and G228A, do not affect telomerase activity, and individuals carrying these sequence alterations have normal telomere length independent of the presence of BMF.
The existence of large deletions and in vitro transfection studies led to the conclusion that haploinsufficiency of TERC is the mechanism underlying AD DC.
TERT Mutations.
Heterozygosity for nonsynonymous mutations in the TERT gene have been identified in individuals with aplastic anemia and short telomeres and also in individuals in whom pulmonary fibrosis was originally diagnosed. A number of such mutations identified are shown in Figure 7-12, D . Mutations located in the N-terminal critical for binding of TERC RNA and within the central region of TERT defining the catalytic region of the reverse transcriptase impair telomerase activity to a variable degree. Mutations in the C-terminal region affect in vivo telomere synthesis after the assembly of a catalytically active enzyme. Disease manifestations in families with TERT gene mutations are highly variable, and frequently disease does not segregate with the mutation in these families, indicating incomplete disease penetrance. No correlation has emerged between site of mutation and disease manifestations, but severity does increase in succeeding generations due to anticipation (see Box 7-11 ). Thus earlier generations may present as adults with pulmonary fibrosis or liver cirrhosis, whereas later generations may present as children with aplastic anemia or dyskeratosis congenita. Interestingly, individuals with a de novo TERT gene deletion secondary to a large chromosomal deletion on chromosome 5p (5p− syndrome, cri du chat syndrome) show accelerated telomere shortening but no clinical features characteristic of DC. In contrast, biallelic TERT gene mutations have been identified in patients with classic manifestations of DC and in one individual with HHS, which suggests that the mutant alleles are expressed codominantly and the residual telomerase activity determines the clinical phenotype. Biallelic TERT or TERC null mutations have never been described, suggesting that in contrast to mice, complete lack of telomerase activity is not compatible with life in humans.
Genetic anticipation occurs when an inherited disease develops at increasingly younger ages or with increased severity in each succeeding generation. Thus if the disease develops in the offspring of a patient, it will tend to do so at an earlier age and display more severe clinical manifestations than in the parent. Disease anticipation is found in patients with autosomal dominant dyskeratosis congenita and is caused by the inheritance of increasingly shorter telomeres in subsequent generations.
TINF2 Mutations.
TINF2 mutations causing DC are strikingly clustered in the TIN2 protein (see Fig. 7-12, E ). Two thirds of the mutations affect a single amino acid residue, Arg282, changing it to a histidine or cysteine residue. Nearly all of the other identified mutations affect the same small region of the protein, encoded in exon 6. In vitro experiments have shown that TIN2, along with TPP1, is involved in recruitment of telomerase to telomeres, and overexpression of the pathogenic TINF2 mutants disrupts the association between TIN2 and telomerase. Intriguingly the cluster of mutations in TIN2 overlaps with the HP1-gamma binding site in the protein that is essential for cohesion, the physical association of newly replicated telomeres. It has been speculated that telomere cohesion may be necessary for efficient telomere elongation by the dimeric telomerase enzyme.
TCAB1 Mutations.
Two unrelated cases of DC caused by TCAB mutations have been reported. In both families the probands were compound heterozygotes for single point mutations causing amino acid substitutions, F164L and R398 in one case and H367Y and G435R in the other. All the affected amino acids are highly conserved. The mutations result in decreased accumulation of TCAB1 and its mislocalization in the cytoplasm rather than the Cajal body. This causes accumulation of TERC in the nucleolus rather than the Cajal body, which presumably affects the subsequent trafficking of telomerase to the telomere. In keeping with this premise, iPSC from TCAB1 DC patients shows normal levels of telomerase activity but defects in telomere lengthening.
CTC1 Mutations.
Most patients are compound heterozygotes, and the mutations include missense, nonsense, frameshift, and inframe deletions and splicing mutations. CTC1 mutations have also been implicated in a rare syndrome known as Coats plus syndrome or cerebroretinal microangiopathy with calcifications and cysts. Null mutations are never found on both alleles of a single patient, suggesting that some CTC1 activity is essential for life. The pathogenetic mechanism by which these mutations cause a DC-like phenotype is unknown. Telomere length is normal or short.
RTEL1 Mutations.
Biallelic missense mutations are the major cause of the severe DC (HHS) caused by RTEL1 mutations, though dominant mutations have been reported. Mutations are mainly missense but include a splicing mutation and a nonsense mutation. An Arg981Trp mutation is recurrent. Examination of patients’ cells using an assay that measures telomeric circles demonstrated that levels of these circles are higher than in controls. RTEL1 is responsible for disassembly of T loops, which are produced during faulty processing of telomere ends and cause telomere shortening. In mice a biallelic Rtel1 deletion is not compatible with life, and no biallelic null mutations have been indentified in humans, suggesting that some RTEL1 function is essential. No defects in homologous recombination, which also involves RTEL1, have been found.
Genotype-Phenotype Correlation
The disease phenotype of patients with DC is highly variable and is influenced by the gene mutated, the pattern of inheritance (X-linked, autosomal dominant, or autosomal recessive), the nature of the mutation, and the history of inheritance (number of generations that the mutations have been passed on to). A distinctive feature of DC is that clinical manifestations differ with the age of disease onset (see Fig. 7-13 ).
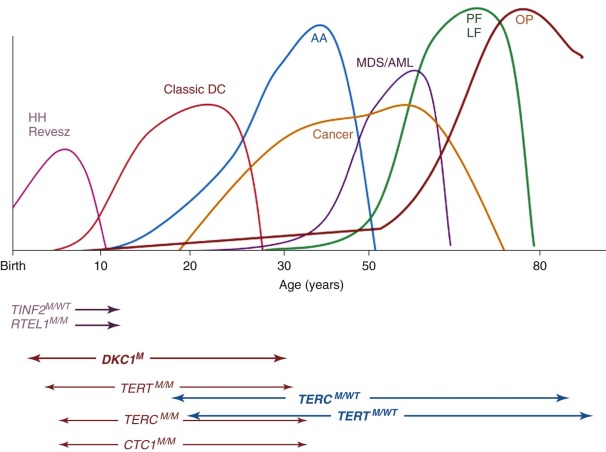
DKC1 Mutations.
The severity of disease in individuals with X-linked DC varies from hardly detectable to a severe form (HHS; see later) that causes death in early childhood (see Fig. 7-12, A ). DKC1 maps to the X chromosome, so the majority of affected individuals are therefore males. The most frequent clinical manifestation is classic DC with mucocutaneous findings in early childhood and progressive BMF in adolescence.
Female carriers of the DKC1 mutation rarely show signs of hematologic disease. Cutaneous manifestations may be found in female carriers but are usually mild. Female carriers of X-linked DC show a phenomenon known as skewed or biased X-inactivation. In females, inactivation of one X chromosome must occur at random early in development to achieve the same dosage of X-linked genes as in male cells. When X-inactivation has been established in a particular cell, it is faithfully passed to all progeny. In women, tissues are therefore a mosaic in which half the cells have the active maternal and half the active paternal X chromosome. In female carriers of DC, blood consists entirely of cells in which the X chromosome containing the normal dyskerin gene is active ; this finding indicates that after random X-inactivation, cells with wild-type dyskerin have a growth or survival advantage (or both) and outgrow mutant cells. Whether this outgrowth takes place in embryogenesis or childhood and whether all tissues are affected are not known. Telomere length in peripheral blood cells in female DC carriers varies from within normal limits to very short. Neither MDS nor MDS/AML has been described in a female DKC1 mutation carrier. Whether female carriers are at increased risk for the development of cancer in other organs remains to be determined. Biased X-inactivation can be used to determine whether DC in a family is X-linked.
TERC Mutations.
The clinical phenotype in patients with AD DC caused by TERC gene mutations is usually mild. Analysis of family members of individuals with AD DC showed that mutation carriers frequently have only subtle signs of BMF but lack the mucocutaneous features of classic DC (atypical DC; see also Fig. 7-7 ). Interestingly, the only patient reported to be a compound heterozygote for two different TERC gene mutations showed signs of classic DC and early onset of disease, whereas family members with only one of the two mutations did not. TERC gene mutations were identified in a subset of adult patients in whom familial or acquired idiopathic aplastic anemia was originally diagnosed and in patients in whom idiopathic pulmonary fibrosis was originally diagnosed. Investigation of family members revealed that in all patients the TERC gene mutations are inherited and that all mutation carriers have short (<10th percentile) or very short (<1st percentile) telomeres. However, short and very short telomeres were additionally found in individual family members who did not carry the mutation and had no signs of disease. TERC gene mutations were likewise identified in some patients with cytopenic MDS or MDS/AML. The mutations identified were associated with reduced telomerase activity in vitro and very short telomeres in the mutation carriers with BMF. The presence of TERC gene mutations in patients in whom pulmonary fibrosis, aplastic anemia, MDS, or MDS/AML was originally diagnosed indicates that the underlying problem in these patients is the pathology of DC and that the variable expressivity and penetrance of the clinical manifestations in DC lead to a much broader clinical picture than originally anticipated when investigating only individuals with classic DC. Interestingly, TERC gene alterations were rarely found in children with aplastic anemia or MDS in the absence of a family history, consistent with the later onset of disease in individuals with AD DC. Pulmonary fibrosis and liver cirrhosis were frequent and sometimes even the first clinical finding in individuals or families with DC and TERC gene mutations.
Anticipation.
AD DC is one of a small number of diseases that show genetic anticipation (i.e., the disease becomes more severe and has an earlier age at onset in later generations). Anticipation became apparent when asymptomatic parents of affected individuals were found to also have TERC gene mutations (see Box 7-11 ). In several cases, examination of these asymptomatic parents revealed that they had signs of mild anemia, such as raised mean corpuscular volume. In other pedigrees, anemia develops in the parents at an older age than in the children. The mechanism of anticipation appears to be that telomere shortening increases in later generations carrying a defective TERC allele. Figure 7-14 shows the inheritance of preshortened telomeres in a nuclear family with AD DC caused by a TERC gene deletion. Thus AD DC develops if an individual inherits a defective TERC or TERT allele and shortened telomeres. It is not known how many generations are needed to produce telomeres short enough to cause the disease. Studies in individuals with a de novo deletion of 5p that includes the TERT gene (5p− syndrome) indicate that in the first generation with TERT haploinsufficiency, telomere shortening is minimal. Interestingly, in individuals with DC and BMF, the telomeres are all equally short, suggesting that there is a minimal length of telomeres required for a progenitor cell to contribute to the peripheral blood cell pool.
TERT Mutations.
Nonsynonymous mutations, small deletions, and splice-site mutations in the telomerase reverse transcriptase TERT gene have been identified in individuals in whom aplastic anemia with short telomeres was diagnosed and in individuals in whom pulmonary fibrosis was diagnosed, indicating that TERT gene mutations may account for cases of atypical DC. Anticipation has also been described in families with AD DC secondary to TERT gene mutations. Individuals with heterozygous TERT gene mutations rarely have the classic form of DC; the majority have atypical DC. Pulmonary fibrosis is a frequent finding in patients and families with atypical DC caused by TERT gene mutations. Mutation carriers with no signs of disease (silent DC) are often found in families with heterozygous TERT gene mutations. Whether telomere length may identify silent TERT gene mutation carriers remains to be determined. Although one would expect that the disease in AD DC secondary to TERC or TERT mutations would be similar, initial family studies indicate that penetrance and expressivity differ in families heterozygous for TERC or TERT gene mutations. The most likely explanation is that TERC or TERT is limiting for different tissues and that expression of a functional TERT protein is regulated differently from regulation of TERC gene expression. Biallelic TERT gene mutations may lead to the classic manifestations of DC and have also been identified in one individual with HHS, which suggests that the mutant alleles are expressed codominantly and that residual telomerase activity determines the severity of the clinical phenotype. Biallelic null mutations of TERT or TERC have never been described, suggesting that in contrast to mice, in humans complete lack of telomerase activity is incompatible with life.
NOP10 and NHP2 Mutations.
The one family with DC due to a homozygous R34W mutation in NOP10 contained three affected children who all showed the classic mucocutaneous features of DC. Only one of the children had BMF. Affected individuals had very short telomeres and low TERC levels, whereas heterozygous family members were asymptomatic but did have shorter than normal telomeres and lower levels of TERC than healthy controls. Whether the lack of BMF in two of the three affected children is due to different genetic backgrounds or mosaicism due to mutations in bone marrow cells is not known. Two individuals with DC due to biallelic mutations in NHP2 have been reported. One, who was a homozygote, had nail dystrophy, thrombocytopenia, and mental retardation, while the other, a compound heterozygote, followed a classic DC course with pancytopenia at 12 years.
TINF2 Mutations.
Patients with TINF2 mutations are amongst the most severely affected of all DC patients. Most patients present in infancy with very short telomeres and often with features of HHS or Revesz syndrome (retinopathy as well as pancytopenia, cerebellar hypoplasia, and developmental delay). TINF2 mutations are dominant in that only one allele is mutated, but they are generally sporadic in that they are due to de novo mutations, patients being so severely affected that they do not live to reproduce (see also Fig. 7-12, E ).
TCAB1 Mutations.
Two unrelated cases of DC due to compound heterozygous TCAB1 mutations have been reported. Both patients had a classic DC presentation, with the mucocutaneous triad and BMF.
CTC1 Mutations.
Patients with CTC1 mutations present with a variety of abnormalities, which include many of the characteristic DC features. CTC1 mutations are also found in patients diagnosed with Coats plus syndrome or cerebroretinal microangiopathy with calcifications and cysts. This is a rare multisystem disorder characterized by progressive intracranial calcifications and leukoencephalopathy, often with the development of intracranial cysts. Retinopathy, BMF, and, less frequently, nail dystrophy, graying or sparse hair, and skin pigmentation abnormalities are also seen. Many of these features overlap with those seen in patients with DC, and CTC1 mutations have in fact been found in patients diagnosed with DC. Patients included those with BMF or some of the mucocutaneous DC features with presence or absence of intracranial calcifications and retinopathy. Telomere length has been reported as short or normal.
RTEL1 Mutations.
A group of seven families with biallelic mutations in RTEL1 was reported by Walne and colleagues. These patients had a consistent type of HHS, characterized by BMF, cerebellar hypoplasia, microcephaly, intrauterine and extrauterine growth retardation, B-cell immunodeficiency, and early death. Abnormal skin pigmentation was absent, and less than half the patients had nail dystrophy or leukoplakia. Le Guen and colleagues reported on three patients from two families with HHS. Affected children in both families had compound heterozygosity for an RTEL1 missense mutation. Fibroblasts from children with RTEL1 gene deletions showed telomere shortening, increased spontaneous 53BP1 foci formation, anaphase bridges, sister telomere losses, and terminal deletions. Ballew and associates described three families with RTEL1 mutations. One kindred had HHS due to biallelic RTEL1 mutations. The other two families harbored heterozygous RTEL1 mutations. In one family, two children with HHS inherited an R1010X nonsense mutation from their healthy mother. The other patient was a man in his 20s who was diagnosed with DC-like disease and had an A645T mutation. Thus it seems that biallelic RTEL1 mutations are a common cause of HHS, and time will tell how frequently dominant mutations are found in BMF syndromes.
Molecular Diagnosis of Dyskeratosis Congenita
Currently, molecular diagnostic tests are not routinely performed as the first line of investigation for DC because they are expensive, labor intensive, and informative in only half of patients. Nevertheless, identification of pathogenic mutations in one of the DC genes is the only way to definitively establish the diagnosis of DC. Furthermore, molecular diagnostic tests enable the identification of mutation carriers, facilitate prenatal diagnosis for an existing pregnancy, and allow preimplantation genetic diagnosis for partners identified as mutation carriers. In addition, because the clinical manifestations, severity of disease, prognosis, and response to treatment may vary, depending on the affected gene and the nature of the individual mutation, molecular diagnostic tests may also influence medical management not only for the individual carrying the mutation but also for unaffected family members. Currently, an educated candidate gene approach, in which the gender ages at disease onset and family history of the affected individual are taken into account, is used to identify the affected gene (see also Fig. 7-13 ). In an affected male, after exclusion of the most frequent DKC1 mutation (A353V), it might be necessary to perform a mutational screen of the entire coding sequence of all three DC genes (usually only TER C and TERT in an affected female). For TERC and TERT the screening methods should include techniques that will detect large deletions. In addition to the large number of mutations in DC genes associated with DC, a number of nonpathogenic mutations and mutations of unknown pathogenic significance have been identified. Determining the pathogenicity of a particular mutation may be difficult.
Telomere measurement in peripheral blood cells and peripheral blood lymphocytes has emerged as a sensitive screening tool for DC in patients with BMF. Whether telomere measurements performed on peripheral blood cells may also identify DC mutation carriers or individuals with DC but nonhematopoietic disease manifestations is controversial.
Diseases Related to Dyskeratosis Congenita
Idiopathic Aplastic Anemia.
A proportion of patients with idiopathic aplastic anemia or hypoplastic MDS were found to have mutations in TERC or TERT. These patients therefore have AD DC. The prevalence of TERC mutations among individuals in whom idiopathic aplastic anemia or hypoplastic MDS is diagnosed appears to be between 2% and 10%. TERC mutations are rare in children with severe aplastic anemia or MDS consistent with later onset of the AD disease. Because parents are often asymptomatic as a result of the anticipation phenomenon and because of incomplete penetrance of TERC and, in particular, TERT gene mutations, a family history is often not apparent and a diagnosis of idiopathic aplastic anemia or idiopathic MDS is made. As a consequence patients below the age of 50 years with acquired aplastic anemia or hypoplastic MDS are now routinely screened for telomere length to identify those who have BMF due to DC. Due to natural telomere shortening with age, telomere length measurements are less sensitive for the identification of patients with underlying DC mutations who are older than 50 years.
Hereditary Pulmonary Fibrosis.
Pulmonary fibrosis, especially in patients with AD DC, may be an initial and sometimes prominent feature of those with atypical DC. Families with hereditary pulmonary fibrosis of adult onset should be investigated for an underlying TERT or TERC gene mutation. The exact pathogenesis of pulmonary fibrosis in patients with DC is not understood. Families with pulmonary fibrosis and aplastic anemia almost always have an underlying TERT or TERC gene mutation. Interestingly, pulmonary fibrosis is frequently found in the older generation, whereas aplastic anemia manifests earlier in life and manifests in subsequent generations (see also Fig. 7-8 ). Thus pulmonary fibrosis in the 50s in the first generation and aplastic anemia in the 20s not only illustrates disease anticipation but also the unique features of age-specific disease manifestation characteristic for AD DC (see Figs. 7-8 and 7-13 ).
Hoyeraal-Hreidarsson Syndrome.
Hoyeraal-Hreidarsson syndrome (HHS; MIM 300240) is a rare, severe variant of DC that presents in early childhood. It is characterized by intrauterine growth retardation, microcephaly, cerebellar hypoplasia, mental retardation, progressive combined immune deficiency, and aplastic anemia. Delayed myelination and hypoplasia of the corpus callosum have been described in patients with HHS. HHS may be caused by a mutation in the DKC1 gene (see Fig. 7-12, A ). It is also notable that several cases of HHS are due to the recurrent mutation (A353V) that is responsible for a third of the X-linked DKC1 cases. In addition, HHS has been found in one individual autozygous (homozygosity in a consanguineous family) or compound heterozygous for a TERT gene mutation and in an individual heterozygous for a TERT gene mutation and anticipation, suggesting that HHS may also be a consequence of a severe telomerase defect alone. In fact, most, although not all, of the HHS patients have very short telomeres at the time of diagnosis. Recently biallelic mutations in RTEL1 have been identified as responsible for a proportion of patients with HHS with short telomeres.
Revesz Syndrome.
The association of progressive bilateral exudative retinopathy (Coats retinopathy), intrauterine growth retardation, fine sparse hair, fine reticulate skin pigmentation, ataxia secondary to cerebellar hypoplasia, cerebral calcifications, extensor hypertonia, and progressive psychomotor retardation with progressive BMF and death in early childhood is also known as Revesz syndrome (MIM 268130), another severe form of DC. Similar to patients with HHS, individuals with Revesz syndrome have very short telomeres. TINF2 exon 6 mutations have been identified in patients with Revesz syndrome.
Ataxia-Pancytopenia Syndrome.
Ataxia-pancytopenia syndrome is a BMF syndrome originally described with several similarities to severe forms of DC. In patients with this syndrome, BMF is associated with severe cerebellar hypoplasia and chromosomal instability without an increase after DEB or MMC testing. MDS and MDS/AML with monosomy 7 are often associated with this syndrome. A TINF2 mutation and short telomeres were identified in one patient, suggesting that the ataxia-pancytopenia syndrome patients have severe DC, HHS, or Revez syndrome.
Coats Plus Syndrome.
Coats plus syndrome (MIM 612199), or cerebroretinal microangiopathy with calcifications and cysts, is caused by compound heterozygous mutations in CTC1 . Many features of these related disorders overlap with those of DC. These features include hair, skin, and nail changes; anemia; thrombocytopenia; osteopenia; intracranial calcifications; exudative retinopathy; and developmental delay. Telomere length in individuals with CTC1 mutations are more variable.
Poikiloderma with Neutropenia.
Poikiloderma with neutropenia (Clericuzio-type poikiloderma with neutropenia, MIM 604173) is a rare AR disease characterized by early-onset poikiloderma, pachyonychia, palmoplantar hyperkeratosis, skeletal defects, and noncyclic neutropenia. The disease is caused by mutations n the USB1 gene (alias C16orf57 and MPN1) and shares some of the clinical characteristics with DC, including abnormal skin pigmentation (poikiloderma, seen in all of the patients), nail dystrophy, and neutropenia and sometimes other characteristics such as leukoplakia, epiphora, short stature, and pulmonary problems. USB1 encodes a protein that is essential for the processing and stability of U6 snRNA, a molecule with a crucial role in RNA splicing; whether USB1 also plays a role in telomere biology remains to be determined. Patients with poikiloderma with neutropenia have normal telomere lengths in their circulating blood cells. It is therefore controversial as to whether poikiloderma with neutropenia should be included in the group of patients classified as having DC or rather represents a distinct disease category. Patients with poikiloderma with neutropenia have a predisposition to developing MDS and squamous cell carcinoma of the skin.
Animal Models.
Dyskerin is highly conserved throughout evolution in its role in rRNA modification and processing but only interacts with telomerase in vertebrates. Thus yeast mutants of Cbf5 show defects in rRNA processing, ribosome biogenesis, and growth; null mutations are lethal. The dyskerin orthologue in Drosophila is Nop60B, also known as minifly, and null mutations are again lethal, while partial loss of functional mutations cause pleiotropic effects, including extreme reduction in body size, developmental delay, and reduced female fertility. These effects have similarities to minute mutants in Drosophila , which are caused by ribosomal protein mutations. They are also similar to features of DC, strengthening the idea that ribosomal defects may contribute to the spectrum of abnormalities in DC. In zebrafish, knockdown or mutagenesis of dyskerin or other H/ACA snRNP proteins (GAR1 or NOP10) leads to defects in ribosome biogenesis and p53-dependent inhibition of hematopoiesis without affecting telomerase activity, making extrapolation to the human disease difficult.
In mice, models of X-linked DC have been hampered by the fact that laboratory mice have very long telomeres that do not become critically shortened in the presence of a compromised dyskerin protein. Nevertheless mutations that are copies of human DKC1 mutations do cause increased DNA damage and defects in hematopoietic stem cell repopulation that are partially corrected by treatment with the antioxidant N-acetyl cysteine. The most promising mouse models have involved deficiency of telomerase itself or defects in components of shelterin. Laboratory mice can thrive without either TERT or TERC, but after several generations of breeding, telomeres shorten and eventually mice show many of the features of DC, notably degenerative effects in tissues with high turnover such as hematopoietic cells, testicular cells, and cells of the skin and gastrointestinal tract. In wild CAST/EiJ mice, which have shorter telomeres, haploinsufficiency for Tert and Terc genes causes DC-like features in early generations. Defects similar to those in DC are also seen in mice lacking shelterin component Pot1b in combination with reduced telomerase activity. These mice show rapid telomere shortening, skin and nail defects, and fatal BMF. Pot1b is a variant form of POT1 found in mice but not humans. These findings illustrate that the mouse can faithfully model telomere diseases and demonstrate the importance of dysfunctional telomeres in DC pathogenesis.
iPS Cells.
iPS cells are pluripotent cells derived from differentiated cells such as fibroblasts by reprogramming with a cocktail of transcription factors. They have been heralded as having the potential for tissue renewal and regeneration and are immediately useful as models for inherited diseases as they carry the patient’s genome and can be cultured to undergo differentiation into many mature cell types. iPS cells have been isolated from DC patients with disease caused by DKC1 , TERC , hTERT, and TCAB1 mutations. The efficiency of reprogramming was low compared with that found in other diseases, perhaps reflecting the fact that stem cell defects are one of the cardinal features of DC. In contrast to other cell lines obtained from patients with DC, iPS cell lines express telomerase activity and therefore allow the study of telomere dynamics in the disease that cannot be studied otherwise. Whether undifferentiated iPS cell lines derived in DC are able to maintain their telomeres over time is controversial and awaits further studies. Nevertheless iPS cells from DC patients are promising, powerful, and unique tools for studying human telomere biology and the molecular pathogenesis of DC. However, there is likely a long path before iPS cell technology fulfills its potential in tissue regenerative therapeutic approaches, the goal that makes this technology so attractive in BM research.
Mechanism of Disease Pathogenesis in Dyskeratosis Congenita
Dysfunctional telomeres are the likely central theme in the pathogenesis of DC. Figure 7-11 summarizes our current model of the pathogenesis of disease in DC.
Clinical Management
Most of what we know regarding therapy and prognosis comes from our experience with patients with classic DC. Care must be taken when extrapolating such experience to all DC patients. The prognosis for patients with classic or severe DC is usually poor. In males with X-linked and sporadic DC, the median age at death was 20 years. Patients with AD DC have milder disease with a better survival rate. Deaths were usually due to complications from aplastic anemia, BMT, pulmonary fibrosis, or cancer.
Only identification of a pathogenetic mutation can confirm the diagnosis. Once confirmed, the family should be referred to a clinical geneticist familiar with new developments in DC for counseling and careful examination of family members. Genetic testing and possibly telomere measurement should be offered to all family members. Early diagnosis is important for correct management of hematologic complications; appropriate treatment of associated pulmonary, gastrointestinal, ocular, dental, and bone disease; identification of affected presymptomatic family members and unaffected family members or pregnancies that may serve as potential hematopoietic stem cell donors; and targeted cancer surveillance, not only in the patient with DC but also in mutation carriers.
Unfortunately, there is currently no consensus on how to manage patients with DC. Management depends on the age at diagnosis, the presence or absence of clinical signs, the gene affected, the molecular characteristic of the mutation identified, and possibly the telomere length. All patients with DC and hematologic manifestations should have a full hematologic assessment, including examination of bone marrow, and in anticipation of possible HSCT, HLA typing should be performed. Other investigations at diagnosis should include pulmonary evaluation and examination of the oropharynx and anal region; in females, cytologic examination of the vagina and cervical epithelium is needed. The prognosis and life expectancy of family members who have inherited only the short telomeres but not the gene mutation are unknown. Whether these family members should be excluded as potential sibling donors of an affected family member is also unknown.
Management of Hematologic Disease
Treatment of BMF in patients with classic DC is similar to that in patients with FA. Initially, in the absence of hematologic abnormalities or clinical and laboratory signs of mild BMF, monitoring of peripheral blood values might be sufficient. In the presence of hematologic abnormalities, yearly bone marrow examination should include a trephine biopsy and marrow aspiration for the examination of marrow cellularity and morphologic changes characteristic of MDS, as well as for cytogenetic examination by G banding and FISH analysis; testing should include probes that identify the most frequent cytogenetic abnormalities seen in patients with DC and progression to MDS or AML, or both (see earlier also). Plans for possible HSCT should be in place because hematologic complications may develop rapidly. With progression of BMF it will become necessary to increase the frequency of hematologic monitoring, and it may have to be individualized. The anabolic steroid oxymetholone can produce improvement in hematopoietic function. Approximately two thirds of patients with DC will respond to oxymetholone; in some cases this response can last several years and involve all lineages. Patients with DC may respond to a dose as low as 0.25 mg of oxymetholone/kg/day, which can be increased, if necessary, to 2 to 5 mg/kg/day. Hematopoietic growth factors usually, if at all, have only a transient effect and should not be used in combination with androgens because of the risk of peliosis and splenic rupture. Unlike idiopathic aplastic anemia, immunosuppressive agents such as antithymocyte globulin and cyclosporine are ineffective or only have a transient response in cases of DC and BMF.
The ultimate treatment of BMF is allogeneic HSCT. However, HSCT for DC has been associated with high morbidity and mortality rates, more so than in other BMF syndromes. Pulmonary and vascular complications are the major causes of death associated with HSCT in patients with DC. Reduced-intensity nonmyeloablative fludarabine-based conditioning regimens with no or minimal irradiation are associated with lower toxicity and fewer complications. Long-term survival and late complication rates after HSCT in patients with DC remain to be determined.
Management of DC patients with MDS or AML (or both) is even less standardized. The significance of transient clonal cytogenetic abnormalities is unclear, but if persistent or complex, such abnormalities usually predict a poor prognosis. Success is unlikely with early hematopoietic stem cell harvest for autologous transplantation at a later time because of the limited repopulating ability of the affected stem cells.
Management of Pulmonary Disease
There is currently no treatment of pulmonary disease in DC. Cigarette smoke is known to accelerate disease onset and severity. Thus patients with DC should refrain from smoking, avoid drugs with pulmonary toxicity (e.g., bleomycin), and have their lungs shielded from irradiation during BMT or cancer treatment. Pulmonary fibrosis in patients with DC does not respond to immunosuppressive therapy. Lung transplantation might be considered in individuals with otherwise mild manifestations of disease.
Cancer Surveillance and Treatment
Patients with confirmed DC should avoid excessive exposure to ultraviolet light and use barrier sun creams. Avoidance of smoking and alcohol should be emphasized, particularly in patients with oral leukoplakia. DC patients should also undergo regular dental and hygienist review to prevent early tooth loss and reduce the occurrence of leukoplakia. It is important to screen for malignant disease, especially of the gastrointestinal system. Management of malignant disease may be complicated by a number of factors. The diagnosis may be delayed in some cases because of malignant change occurring within an area of persistent leukoplakia, resulting in a delay in treatment. There is also a high potential for local recurrence and multiple cancers in these patients. In addition, the presence of BMF makes chemotherapy very difficult and can limit the indications for surgery. The mucosa also has high sensitivity to irradiation, which can lead to severe mucosal damage and may result in early termination of radiotherapy.
Management of oral malignant disease usually includes a regular review and biopsy of the premalignant hyperkeratotic lesion and surgical laser excision of any suspicious areas. Neoadjuvant radiotherapy or surgical resection of confirmed malignant tumors, along with ipsilateral node clearance with or without adjuvant radiotherapy in accordance with staging of the disease, is the mainstay of treatment.
Gene Therapy
HSCT may improve or cure hematologic disease in DC patients, but HLA-matched sibling donor cells are available for only a minority of patients; furthermore, immune reconstitution is often delayed. Gene therapy to correct hematopoietic stem cells followed by autologous transplantation might offer an attractive alternative for the treatment of hematologic disease in DC patients. Biased X-inactivation in female DKC1 mutation carriers suggests that DC cells corrected by gene therapy are likely to have a selective growth and survival advantage. Indeed, gene transfer of TERT or TERC (or both) corrects the telomere defect in most DC cell lines. However, as in many other IBMFSs, the few surviving hematopoietic stem cells might have already acquired secondary somatic and possibly oncogenic mutations, so correction by gene therapy could promote rather than prevent leukemogenesis in these patients. Interestingly, in rare patients with DC due to TERC gene mutations, gene reversion by mitotic recombination in hematopoietic stem cells leads to two normal copies of the TERC genes in the majority of circulating blood cells. Revertant cells contribute to the myeloid and B lymphocyte lineages but were not detectable in T lymphocytes, suggesting that mitotic recombination occurred in a stem cell committed to myeloid and B-cell differentiation or that the selective advantage for gene revertant cells is less in T cells than in myeloid and B cells. Indeed, T cells are less severely affected by DC mutations (see also above) and have a longer survival time in circulation. Revertant mosaicism in DC seems to be less frequent than reported in FA and currently has only been described in DC patients with TERC gene mutation. However, as in FA, DC revertant mosaicism might mask an inherited TERC gene mutation when genetic testing is performed in peripheral blood cells. DNA from skin fibroblasts or buccal swabs might have to be sent for genetic testing in patients with classic DC features but no BMF. Revertant mosaicism in patients with DC but no BMF powerfully illustrates the feasibility of gene therapy and shows that the restoration of a normal TERC gene in a single hematopoietic stem cell in these patients can restore blood cell production.
Shwachman-Diamond Syndrome
Shwachman-Diamond syndrome (SDS, MIM 260400) is a rare, recessive multisystem disorder characterized by exocrine pancreatic insufficiency, bone marrow dysfunction, and skeletal abnormalities, first described in the 1960s ( Box 7-12 ).
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


