Fig. 7.1
Model of p53 stabilization in response to impaired ribosome biogenesis in DBA. Normal erythroblasts produce large numbers of ribosomes for protein synthesis. Levels of p53 remain low via a feedback loop whereby MDM2, a transcriptional p53 target, ubiquitinates p53 to promote its degradation by proteasomes. However, haploinsufficiency for specific RPs causes accumulation of other RPs, which bind to MDM2, thereby inhibiting its ability to promote p53 degradation. Consequently, p53 accumulates and triggers cell cycle arrest and apoptosis
7.1.1.4 Clinical Management
Clinical Presentation
Anemia
DBA is a congenital pure red cell aplasia characterized by normochromic macrocytic anemia and reticulocytopenia in the peripheral blood and the loss of erythroblast from bone marrow. Approximately 90% of affected individuals present in infancy or early childhood, although a “nonclassical” mild phenotype may not be diagnosed until later in life [2, 3].
Congenital Anomalies
Clinical manifestations of DBA are variable. In about 40% of patients, DBA is associated with physical anomalies and growth retardation, but in some patients, no congenital anomalies are found [3]. Table 7.1 summarizes the range of congenital abnormalities found in DBA patients.
Table 7.1
Congenital abnormalities observed in Diamond-Blackfan anemia (DBA)
Head, face, palate | Hypertelorism, cleft palate, high-arched palate, microcephaly, micrognathia, low-set ears, low hairline, epicanthus, ptosis, etc |
Upper limb | Triphalangeal, duplex or bifid, hypoplastic thumb; flat thenar eminence; syndactyly; absent radial artery |
Renal, urogenital | Absent kidney, horseshoe kidney, hypospadias |
Cardiopulmonary | Ventricular septal defect, atrial septal defect, coarctation of the aorta, complex cardiac anomalies |
Other | |
Neck | Short neck, webbed neck |
Eyes | Congenital glaucoma, strabismus, congenital cataract |
Neuromotor | Learning difficulties |
Short stature | |
Cancer Predisposition
Twenty-nine cases were reported among more than 700 patients in the literature [3]. A recent prospective study showed that among 608 patients, 15 solid tumors, two acute myeloid leukemias (AML), and two cases of myelodysplastic syndrome (MDS) were diagnosed at a median age of 41 years in patients who had not received a bone marrow transplant. The rate of development of solid tumors or leukemia was 5.4-fold higher in DBA patients than expected in a demographically matched comparison with the general population. Furthermore, specific tumors had significantly elevated incidence ratios for MDS, AML, adenocarcinoma of the colon, osteogenic sarcoma, and female genital cancer [138].
Diagnostic Procedures
Diagnostic Criteria
The diagnostic criteria for “classical” DBA include macrocytic (or normocytic) anemia with no other significant cytopenia presenting prior to the first birthday, reticulocytopenia, and normal marrow cellularity with a paucity of erythroid precursors, and it is accompanied by congenital anomalies in some cases. However, diagnosis of DBA is often difficult due to the incompleteness of the phenotypes and the wide variability in clinical expression. Even mutations in individual RP genes lead to widely variable phenotypic expression; family members with the same mutation in an RP gene can present with clinical differences. For example, RPS19 mutations are found in some first-degree relatives presenting only with isolated high erythrocyte adenosine deaminase activity and/or macrocytosis [24]. Therefore, it is very difficult to make a diagnosis on the basis of clinical phenotype alone. Molecular diagnosis enables the detection of carriers and the avoidance of hematopoietic stem cell transplantation from sibling donors with these mutations. However, about 40% have no known pathogenic mutations. The diagnostic and supporting criteria for diagnosis of DBA, including mild type, are described in Table 7.2. We have modified the criteria from the international clinical consensus conference [3] based on our results for the novel DBA biomarker reduced glutathione (GSH) [32].
Table 7.2
Diagnostic criteria for patients with Diamond-Blackfan anemia (DBA)
Diagnostic criteria: |
• Age less than 1 year |
• Macrocytic anemia with no other significant cytopenias |
• Reticulocytopenia |
• Normal marrow cellularity with paucity of erythroid precursors |
Supporting criteria: |
Major: |
• Gene mutation described in “classical” DBA |
• Positive family history |
Minor: |
• Elevated erythrocyte adenosine deaminase (eADA) activity and erythrocyte-reduced glutathione (eGSH)a |
• Congenital anomalies described in “classical” DBA |
• Elevated HbF |
• No evidence of other inherited bone marrow failure syndromes |
“Classical” DBA fulfills all four diagnostic criteria |
“Nonclassical” DBA fulfills one of the following: |
1. Three of the diagnostic criteria and one major or two minor supporting criteria |
2. Two of the diagnostic criteria and three minor supporting criteria |
3. Two major supporting criteria |
Differential Diagnosis
In children, transient erythrocytopenia in childhood (TEC) should be a major consideration [3]. TEC usually presents in children older than 1 year of age following viral infection. Most cases resolve spontaneously within 1–2 months. Patients with TEC present with normocytic anemia, and, in contrast to those with DBA, their HbF levels and erythrocyte adenosine deaminase (eADA) activities are normal. Congenital bone marrow failure syndromes accompanied by physical anomalies include dyskeratosis congenita, Shwachman-Diamond syndrome, congenital amegakaryocytic thrombocytopenia, and Pearson syndrome. Although all of these diseases are extremely rare, it is possible to discriminate them from specific clinical findings. Recently, because the causative genes for all of these diseases have been identified, the molecular etiology may be clarified in the near future, and genetic diagnosis may be possible in many patients.
Treatment
Transfusion Therapy
Patients who do not respond to steroids may require chronic transfusion therapy every ~4–8 weeks. Although maintaining hemoglobin levels above 8 g/dL is recommended, a disadvantage of chronic transfusion therapy is hemosiderosis by iron overload. To avoid liver dysfunction, diabetes, and myocardiopathy due to iron deposition, chelation therapy with deferasirox or deferoxamine is recommended. However, methods for oral iron chelation therapy with deferasirox in neonates have not been established.
Drug Therapy
Approximately 80% of patients respond to steroids. The recommended starting dose is 2 mg/kg/day of prednisone at initial treatment. In over 20% of DBA patients, steroids eventually are stopped completely [3]. It should be mentioned that important steroid side effects include growth retardation, osteoporosis, obesity, hypertension, diabetes mellitus, cataract, and glaucoma. The therapy is not generally recommended in babies under 6 months of age. Alternative treatments, including cyclosporine, metoclopramide, EPO and a combination of prednisolone and cyclosporine, have been mentioned, but evaluations have not been provided.
Hematopoietic Stem Cell Transplantation
Chronic transfusion dependence with refractoriness to steroids is considered an acceptable indication for hematopoietic stem cell transplantation (HSCT). The outcomes of HSCT for DBA in Japan are better than those reported in other countries. To date, 19 patients have received transplants of allogeneic hematopoietic stem cells (HSCs). Thirteen patients underwent BMT (six cases with HLA-matched siblings and seven cases with unrelated donors), and all have disease-free survival [33]. Five patients underwent cord blood transplantation (CBT), and two of these with transplants from siblings have survived. However, in three patients treated with unrelated donor cord blood, two were not successfully engrafted, and the third died of lymphoproliferative disease, despite confirmed engraftment. Consequently, bone marrow should be selected as a source of HSCs for transplantation at present. Better therapeutic outcomes have been observed with a conditioning therapy combining busulfan (oral administration, 16 mg/kg or 560 mg/m2) and cyclophosphamide (120–200 mg/kg). Although the reported number of patients is few, a conditioning therapy using a half dose of busulfan leads to good outcomes when using unrelated HSCs transplantation. When busulfan was omitted from pretreatment, the number of cases with engraftment failure was modestly higher. There is not sufficient evidence supporting the value of bone marrow nondestructive pretreatment therapy for HSCT in DBA [34].
7.1.1.5 Future Challenge
Recent studies have provided fascinating insights into the pathogenesis of DBA. However, several important questions remain to be answered. For example, it is not clear how haploinsufficiency of ribosomal proteins leads to a phenotype of defective erythropoiesis and why mutations in some ribosomal protein genes such as RPL5 have more profound impact on fetal development than mutations in other RP genes. Although corticosteroids remain the mainstay of treatment of DBA more than half a century after the original report of their efficacy, their mechanism of action is still unknown. DBA will necessarily lead us toward a further understanding of bone marrow failure syndrome and potentially to novel ways of treatment.
Recently, using DBA models in zebra fish and mice, it has been reported that administration of l-leucine, an essential amino acid, provided relief from anemia [35, 36]. Clinical trials to evaluate the therapeutic effects of l-leucine are underway in the United States. About half of DBA patients have notbeen identifiedcausative mutations. Comprehensive genetic analysis using next-generation sequencing in the remaining DBA patients would be valuable in the identification of other causative gene mutations.
7.1.2 Fanconi’s Anemia
7.1.2.1 Introduction
Fanconi’s anemia (FA) is characterized by progressive bone marrow failure, congenital abnormalities, and a pronounced susceptibility for malignancy, including myelodysplastic syndrome (MDS), leukemia, and solid tumors such as squamous cell carcinoma. FA was first reported by Dr. Fanconi in 1927 [37]. Lymphocytes from FA patients exhibit chromosomal instability [38], a characteristic that was increased by mitomycin C treatment [39]. Mutations in 19 different genes (FA genes) have been identified, the products of which cooperate with other gene products in a common pathway regulating DNA repair [40]. Stem cell transplantation is the only curative treatment for FA. In recent years, HSCT from alternative donors has become more popular and has been successfully employed [41, 42].
7.1.2.2 Epidemiology
About 5–10 cases are diagnosed with FA, and the incidence in Japan is estimated to be 5 in 1,000,000 newborns. The carrier frequency for FA in Japan may be 1 in 200–300 people [23].
7.1.2.3 Pathogenesis
FA is a multigenic disorder with 19 genes currently identified (A, B, C, D1, D2, E, F, G, I, J, L, M, N, O, P, Q, R, S, and T) [40]. Among them, FANCT was the first FA-causative gene discovered by a Japanese group [43]. FANCD1, FANCJ, and FANCN were identified as BRCA2, BRIP1, and PALB2, respectively. These three FA genes are associated with breast and ovarian cancer in heterozygotes. With the exception of the X-linked FANCB gene, the remaining 18 FA genes are autosomal recessive. The most common gene in FA is FANCA (60–70%), and more than 80% of FA patients have mutations inFANCA or FANCG in Japan. These FA proteins cooperate with other gene products, in a common pathway (FA-BRCA pathway) that regulates DNA repair (Fig. 7.2). The FA pathway plays an important role in the proliferation of hematopoietic stem cells and tumor suppression. The FANCD1 and FANCN subtypes initiate a malignant tumor in early childhood, and their prognosis is very poor. In contrast, clonal expansion of hematopoietic cells with reversion mosaicism can restore normal hematopoiesis [44].
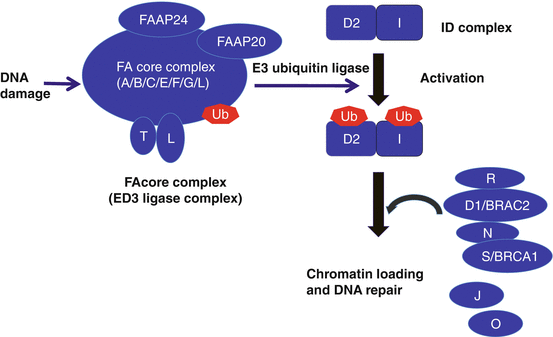
Fig. 7.2
Schematic of the Fanconi’s anemia DNA repair pathway. After DNA damage, the FA core complex is activated, which then functions as an E3 ubiquitin ligase and monoubiquitinates FANCD2 and FANCI. The monoubiquitinated FANCD2/FANCI complex (ID complex) is then targeted to chromatin where it forms a complex with additional FA proteins and other DNA repair proteins
Recent studies in mice have suggested that the FA proteins might counteract aldehyde-induced genotoxicity in hematopoietic stem cells [139, 140]. Acetaldehyde dehydrogenase 2 (ALDH2) primarily catalyzes the conversion of acetaldehyde to acetic acid; it also converts a range of endogenous toxic aldehydes. ALDH2 deficiency resulting from a Glu504Lys substitution (rs671, hereinafter referred to as the A allele) is highly prevalent in East Asian populations, including Japanese. The A allele (Lys504) acts as a dominant negative, since the variant form can suppress the activity of the Glu504 form (G allele) in GA heterozygotes by the formation of heterotetramers [45]. Individuals with the A variant experience flushing when drinking alcohol and have an elevated risk of esophageal cancer with habitual drinking. Compared to individuals with GG homozygotes, enzymatic activity is reduced by 60–80% or nearly abolished (~4%) in GA or AA individuals, respectively [46]. Recently, Hira et al. showed that the presence of the A allele strongly accelerated progression of BMF in Japanese FA patients. Although ALDH2 status did not influence the number of developmental abnormalities, malformations at some specific anatomic locations were observed more frequently in ALDH2-deficient patients [47]. These results indicate that the level of ALDH2 activity impacts the pathogenesis of FA.
7.1.2.4 Clinical Management
Clinical Presentation
Pancytopenia
Pancytopenia was the most common presentation, particularly when the red cell mean cell volume (MCV) and fetal hemoglobin (HbF) were elevated for age. Bone marrow biopsy examination most often showed hypocellularity for age, due to decreased numbers of hematopoietic precursors with normal morphology. The median age at diagnosis is 6.5 years, ranging from birth to adult. The diagnostic age in the reported cases was similar in both sexes [48].
Congenital Anomalies
Clinical manifestations of FA are variable. In about 60% of patients, FA is associated with physical anomalies and growth retardation, but in some patients, no congenital anomalies are found [48]. The most common characteristics at birth are skin hyperpigmentation, café au lait spots, short stature, abnormal thumbs, and radii [49].
Cancer Predisposition
Patients with FA are at a particularly high risk of developing MDS/AML and specific solid tumors at unusually young ages, including head, neck, esophageal, and gynecological squamous cell carcinoma. In the literature, about 15–20% of FA patients had hematologic neoplasms, and 5–10% had nonhematologic neoplasms [50, 51]. Of these neoplasms in Japanese FA patients, 33% were hematologic and 10.4% were nonhematologic [49]. Solid tumors were observed both after and before HSCT, and they developed between 1 and 39 years of age.
Diagnostic Procedures
Diagnostic Evaluation
FA is an autosomal recessive disorder associated with a high frequency of bone marrow failure, leukemia, and solid tumors. FA is a complex disease that can affect many systems of the body. Initially, patients are recognized when they have the combination of aplastic anemia and birth defects. However, current diagnostic criteria are more extensive and rely on demonstration of chromosomal aberrations in cells cultured with DNA cross-linking agents. The suspected diagnosis is usually confirmed by demonstration of chromosomal aberrations in blood lymphocytes cultured with a DNA cross-linking agent such as diepoxybutane (DEB) or mitomycin C (MMC) [48]. Following DNA damage, the complex of upstream FA gene products (A, B, C, E, F, G, I, L, M) leads to ubiquitination of the product of FANCD2, detected with a Western blot with a D2-specific antibody [52].
However, some FA patients have hematopoietic somatic mosaicism. These cases have corrected one mutated allele in a bone marrow stem cell, leading to an acquired heterozygosity in the blood cells. If diagnostic test results of blood are not conclusive and there is a high probability of FA, skin or bone marrow fibroblast cultures are required to demonstrate sensitivity to DNA cross-linking agents. Finally, FA mutation analysis determines the initial complementation group. Target next-generation sequencing can be performed to determine the relevant mutations.
Differential Diagnosis
Chromosome instability syndrome is a group of inherited conditions associated with chromosomal instability and breakage, and the following chromosome instability syndromes are known: xeroderma pigmentosum, ataxia-telangiectasia, Bloom syndrome, and Nijmegen syndrome.
Treatment
Transfusion Therapy
FA patients can be transfused to maintain minimal trough hemoglobin of 6 g/dL and minimal trough platelet of 5000/μL.
Drug Therapy
Patients who choose not to pursue transplant for severe cytopenias may benefit from treatment with androgens. Androgens can improve cytopenias in all three lineages, erythroid, myeloid, and platelets, but the effects are typically most pronounced for the erythroid lineage [48]. The effects of androgens were recognized in half of FA patients, but its effect seems to have lasted only temporarily [53]. Androgen may exacerbate the risk of liver tumors, and prior exposure to androgens is the one of the risk factors affecting survival after unrelated transplant [54]. Metenolone acetate is used in Japan. A clinical trial of danazol, which has less virilizing side effects, is currently under way. The use of steroid hormones should be avoided.
Hematopoietic Stem Cell Transplantation
Currently, the only cure for the hematological abnormalities of FA remains HSCT. The optimal timing of transplantation is challenging because the best outcomes are achieved prior to the development of complications such as infections from chronic severe neutropenia, high transfusion burden to treat anemia/thrombocytopenia, and the development of MDS or AML. Patients with FA are exquisitely sensitive to genotoxic agents such as cyclophosphamide, busulfan, and ionizing radiation. FA patients are also susceptible to the damaging inflammatory side effects of graft-versus-host disease. The use of high-dose cyclophosphamide (CY) and radiation in preparative regimens for FA patients often resulted in excessive organ toxicity and death. Therefore, efforts have focused on reducing the doses required for transplant preparative regimens, choosing nongenotoxic regimens to prevent graft-versus-host disease and using alternative conditioning regimens [48]. The use of low-dose CY (20–40 mg/kg) combined with 4–6 Gy of thoracoabdominal irradiation (TAI) or total body irradiation (TBI) reduced procedure toxicity and improved the outcome for FA patients transplanted from HLA-matched sibling donors [55]. Non-radiation regimens have been increasingly used for FA patients to reduce the secondary malignancies associated with radiation [56, 57].
A significant proportion of FA patients undergoing HSCT can now be dramatically cured, even in the absence of an HLA-identical sibling, especially if the conditioning regimen includes fludarabine [42, 58]. In Japanese FA patients transplanted using a fludarabine regimen, the 3-year estimate of overall survival (OS) for eight patients was 100% when the donor was an HLA-matched sibling [59], and 26 patients out of 27 patients transplanted from an alternative donor are alive [42]. It is essential to test all potential sibling donors for FA regardless of clinical findings since the phenotypic variation even within a given family is broad.
HSCT does not correct the nonhematological manifestations of Fanconi’s anemia. Solid tumor risk, particularly head and neck squamous cell carcinoma, continues to increase after transplant, particularly in FA patients experiencing severe graft-versus-host disease [60]. A retrospective study comparing solid tumor risks in transplanted versus non-transplanted FA patients reported a 4.4-fold higher age-specific hazard rate of squamous cell carcinoma in patients treated by transplantation. The tumors appeared at an earlier age in the transplanted cohort [61]. Data on solid tumor risks in patients transplanted with the newer regimens are as yet limited.
7.1.2.5 Future Challenges
Patients with FA usually are diagnosed in childhood, and their registrations have been managed in the Japanese Society of Pediatric Hematology/Oncology in Japan. Although the adult FA population is small, it is important to understand transplanted/non-transplanted adult FA patients, who have or have not developed bone marrow failure, hematological malignancy, and solid tumors. Fludarabine-based preconditioning regimen can be used satisfactorily in alternative HSCT for FA. However, long-term observation of secondary cancers and other late effects will be required to determine the therapeutic utility of this approach. A recent report showed endogenously produced aldehydes impact the severity of FA, suggesting the possibility of a novel therapeutic approach [47]. For example, Alda-1 can stimulate the enzymatic activity of both the normal and variant ALDH2 [62]. ALDH2 agonists such as Alda-1 might be a novel protective drug against BMF in FA patients.
7.2 TAM
7.2.1 Introduction
Trisomy 21, the genetic hallmark of Down syndrome (DS), is the most frequent human chromosomal abnormality. In Japan, the incidence in the general population is 1 in 600–800 live births. Neonates with DS are at a high risk of developing a hematologic disorder referred to as transient abnormal myelopoiesis (TAM), which is characterized by the rapid growth of abnormal blast cells expressing megakaryocytic markers [63]. It has been estimated that 5–10% of infants with DS exhibit the disorder, and in most cases, it resolves spontaneously within 3 months. However, approximately 20% of severe cases are subject to fatal complications, and 20–30% of patients who escape from early death develop AMKL referred to as myeloid leukemia of DS (ML-DS) within the first 4 years of life [63–66].
Human tumors have been shown to progress by the accumulation of genetic abnormalities [67]. The malignant progression from TAM to AMLK offers a unique model to study the stepwise developments of cancer pathogenesis. The blasts in TAM are indistinguishable from true leukemic cells in both morphology and expressed surface markers. Blast cells in most patients with TAM as well as those with ML-DS have mutations in the gene encoding the transcription factor GATA1 [68–74], which is essential for normal development of erythroid and megakaryocytic cells [75–77]. Recent reports showed that TAM is caused by a single GATA1 mutation and constitutive trisomy 21, and subsequent AMKL evolves from a preexisting TAM clone through the acquisition of additional mutations in genes coding for cohesin components, CCCTC-binding factor (CTCF), epigenetic regulators, as well as tyrosine kinases/Ras pathway genes (Fig. 7.3) [78]. In this chapter, advances in the pathobiology and clinical management of TAM will be described.
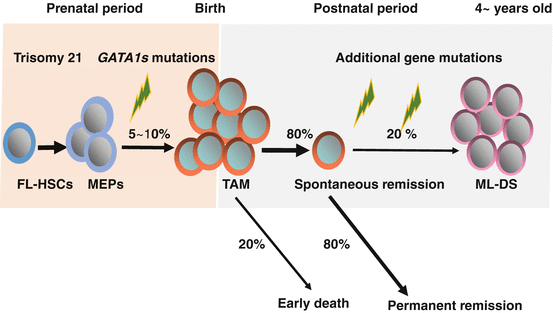
Fig. 7.3
Natural history and pathogenesis of TAM and ML-DS. Trisomy 21, the genetic hallmark of Down syndrome (DS), is the most frequent human chromosomal abnormality. Five to 10% of neonates with DS develop transient abnormal myelopoiesis (TAM), which is characterized by rapid growth of abnormal megakaryoblastic cells. Almost all cases of TAM have mutations in GATA1. In most cases, it resolves spontaneously within 3 months. However, early death occurs in about 20% of the cases. Furthermore, about 20% of patients develop myeloid leukemia of DS (ML-DS) within 4 years of life after acquisition of additional mutations in the genes encoding multiple cohesion components, CTCF, and epigenetic regulations such as EZH2, as well as genes encoding common signaling pathways, such as JAK family kinases and Ras pathway genes. FL-HSCs: Hematopoietic stem cells in fetal livers, MEPs: Megakaryocyte-erythroid progenitors
7.2.2 Epidemiology
The actual incidence of TAM has been difficult to define. Estimates of the frequency of TAM vary from 3.8 to 30% of neonates with DS, depending on the diagnostic and eligibility criteria used in each study and the sensitivity of the methodologies used for screening [66, 79–81]. Zipursky et al. screened a small series of DS neonates by microscopic examination of blood films and found that 10% had blasts in the peripheral blood [66]. It is possible that some cases with relatively few blasts would have been missed. Pine et al. screened Guthrie cards from 590 DS infants for GATA1 mutations by single-strand conformation polymorphisms (SSCP) and direct sequencing of PCR products and showed that 3.8% of the infants harbored GATA1 mutations [80]. A recent prospective study of 200 DS neonates showed an 8.5% frequency of GATA1 mutations, using a combination of standard Sanger sequencing with direct high-performance liquid chromatography (Ss/HPLC) [81]. All of these patients had more that 10% blasts in the peripheral blood. Furthermore, low abundant minor GATA1-mutant clones were detected in 20.4% of DS neonates without detectable GATA1 mutant clones with Ss/HPLC, by more sensitive targeted next-generation sequencing that can detect as few as 0.3% TAM blasts. They referred to these patients as “silent TAM.” This study indicates approximately 30% of DS neonates have GATA1 mutations [81].
7.2.3 Pathogenesis
7.2.3.1 GATA1 Mutations in TAM and ML-DS
Cell differentiation is controlled in part by cell lineage-restricted transcription factors. The transcription factor GATA1 is expressed within the hematopoietic hierarchy in erythroid, megakaryocytic, eosinophilic, and mast cell lineages [82–85], as well as in Sertoli cells of the testis [86, 87]. Gene targeting experiments have revealed that GATA1 is required for the terminal differentiation of definitive erythroid and megakaryocytic cells [88–90].
GATA1 mRNAs are abundantly expressed in blast cells from TAM patients, as well as ML-DS patients [91]. Almost all patients with TAM and ML-DS have GATA1 mutations [68–74]. The mutations lead to a loss of expression of the full-length GATA1 protein and the expression of a truncated GATA1 protein (GATA1s) that lacks the N-terminal transactivation domain. GATA1s, which is translated from the second methionine at codon 84, retains intact zinc finger regions, binds appropriately to the GATA consensus sequence, and interacts normally with its essential cofactor FOG-1.
Several cell culture-based rescue experiments have reported that the N-terminal activation domain appears to be dispensable during erythroid and megakaryocytic cell differentiation [92–95]. Furthermore, distinct regions in the N-terminus of GATA1, which regulates the proliferation of immature megakaryocytic progenitors, are required for terminal megakaryocyte differentiation and controlling the growth of immature precursors [96, 97]. GATA1, but not GATA1s, interacts physically with the transcription factor E2F, which can trigger reentry into the cell cycle. That observation suggests that the failure of GATA1s to repress E2F activity in cellular transformation of fetal progenitors may be caused by this failure in direct or indirect interaction [98]. Supporting this hypothesis, Toki et al. recently discovered novel GATA1 mutants in TAM that lack just the E2F interaction domain in the N-terminus of GATA1 [99].
On the other hand, mice harboring a heterozygous GATA1 knockdown allele frequently develop erythroblastic leukemia [100]. These observations indicate that the expression level of GATA1 is crucial for the proper development of erythroid and megakaryocytic cells and that compromised GATA1 expression is a causal factor in leukemia [101]. Recently, Kanezaki et al. showed that the spectrum of transcripts derived from the mutant genes affects GATA1s protein expression. Mutations resulting in low GATA1s levels are significantly associated with a risk of progression to ML-DS [102]. However, a subsequent report contended that the GATA1 mutational spectrum did not differ between TAM and AMKL and that the type of GATA1 sequence mutation is not a reliable tool and is not prognostic of which patients with TAM are likely to develop ML-DS [103]. To resolve this problem, a prospective study with a large series of TAM patients is necessary.
7.2.3.2 The Target Cells of TAM and ML-DS
TAM is a disorder found mainly in patients with DS during the newborn period and is sometimes associated with liver fibrosis, which usually takes a lethal course. Based on pathological observation of TAM cases with fatal liver fibrosis, Miyauchi et al. have proposed a hypothesis that TAM blasts originate from fetal liver hematopoietic progenitors [104]. Analyses of GATA1 mutations provide direct evidence for this hypothesis. The presence of identical GATA1 mutations in the blasts of both TAM and ML-DS in an identical twin suggested that GATA1 mutations occur early during prenatal hematopoiesis [105]. GATA1 mutations were detected in hematopoietic tissues from DS fetuses that had no pathological evidence of leukemia [106] and in Guthrie newborn screening cards at birth from DS infants who later developed ML-DS [107].
In addition, mice expressing GATA1s presented with hyperproliferation of yolk sac and fetal liver progenitors [108]. Consistent with this finding, GATA1-deficient mice rescued with transgenic expression of GATA1s exhibited hyper-megakaryopoiesis during a limited embryonic and postnatal period, resembling the phenotype in human TAM cases [109]. These data from mouse experiments also indicated the possibility that fetal megakaryocytic progenitors are the target cells of TAM and ML-DS.
7.2.3.3 The Essential Roles of Trisomy 21 in Leukemogenesis of DS
For the following reasons, trisomy 21 is thought to be the first genetic event in the development of ML-DS. First, TAM occurs almost exclusively in patients with DS [66]. Second, when TAM occurs in patients with mosaic trisomy 21, the TAM clones involve only the cells with trisomy 21 [110]. Third, an inherited mutation in humans leading to production of only GATA1s is associated with impaired erythropoiesis and granulopoiesis, but does not promote leukemia in the absence of trisomy 21 [111]. Fourth, trisomy 21 itself is associated with enhanced expansion of human fetal erythroid and megakaryocytic precursors, independent of GATA1 mutations [112, 113].
Related posts:
 Childhood Aplastic Anemia
Childhood Aplastic Anemia
 Hemophagocytic Lymphohistiocytosis, Secondary
Hemophagocytic Lymphohistiocytosis, Secondary
 Pathogenesis and Treatment of Hemophilia
Pathogenesis and Treatment of Hemophilia
 Primary Hemophagocytic Lymphohistiocytosis
Primary Hemophagocytic Lymphohistiocytosis
 Myelodysplastic Syndrome (MDS) and Juvenile Myelomonocytic Leukemia (JMML)
Myelodysplastic Syndrome (MDS) and Juvenile Myelomonocytic Leukemia (JMML)
 Hematopoietic Stem Cells: The Basis of Normal and Malignant Hematopoiesis
Hematopoietic Stem Cells: The Basis of Normal and Malignant Hematopoiesis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


