Inflammation plays a critical role in the complex pathophysiology of sickle cell disease and drives both the acute and chronic processes leading to vascular injury. Mediators of inflammation, such as cellular adhesion molecules, cytokines, leukotrienes, and nuclear factor κB signaling factors, represent potential therapeutic targets in sickle cell disease.
Key points
- •
Inflammation plays a critical role in the complex pathophysiology of sickle cell disease (SCD) and drives both the acute and chronic processes leading to vascular injury.
- •
Mediators of inflammation, such as cellular adhesion molecules, cytokines, leukotrienes (LTs), and nuclear factor (NF)-κB signaling factors, represent potential therapeutic targets in SCD.
Introduction
SCD is now recognized as a complex disease characterized by acute and chronic inflammation. The incidence of nearly every clinical manifestation of SCD correlates with white blood cell (WBC) count, indicating a role for leukocytes and inflammation in the pathophysiology of SCD. Leukocytosis is common in SCD patients and is manifested by elevation in monocyte and neutrophil counts, accompanied by elevated levels of circulating inflammatory cytokines, including tumor necrosis factor α (TNF-α), interleukin (IL)-1, and IL-8. Elevated levels of these cytokines have been shown to exacerbate sickling in experimental models and correlate with clinical vasoocclusive severity. Leukocytosis is associated with a decreased life expectancy and, when observed in infancy, predicts future disease severity. Clinically, elevated baseline WBC counts have been associated with acute chest syndrome (ACS), decline in lung function, and ischemic stroke in children with sickle cell anemia (SCA). Reports of acute vasoocclusive pain events, ACS multiorgan failure, and death after administration of myeloid colony-stimulating factors (granulocyte colony-stimulating factor or granulocyte-macrophage colony-stimulating factor) suggest that the association of leukocytosis and poor outcome may be causal. The reduced incidence of vasoocclusive pain and ACS episodes in patients on hydroxyurea (HU) is thought due, in part, to the myelosuppressive effect of the drug.
Genetic associations with inflammation also support the involvement of inflammatory pathways in the pathophysiology of SCD and may partly explain the phenotypic heterogeneity of the disease. Associations between variants in the transforming growth factor β1 (TGF-β)/bone morphogenetic protein 6 pathway and pulmonary hypertension, stroke, and osteonecrosis in SCD have been replicated in 4 independent studies.
Studies showing an increased risk of severe disease in SCA patients with elevated levels of TNF-α receptor-1 (TNF-R1) and vascular cell adhesion molecule (VCAM)-1 are supported by findings from a genome-wide association study of a link between disease severity and polymorphisms in VCAM-1 and in ADP-ribosylation factor guanine nucleotide-exchange factor 2 ( ARFGEF2) , a gene involved in TNF-R1 release.
Another recent gene-centric association identified an association between ACS and a variant (rs6141803) located in close proximity to a gene ( COMMD7) that is highly expressed in pulmonary endothelial cells (ECs), interacts with NF-κB signaling, and is differentially expressed when exposed to oxidant heme species.
Individuals with SCA who express the integral red blood cell (RBC) membrane glycoprotein, Duffy antigen receptor for chemokines (DARC), were found to have higher steady-state WBC counts and levels of DARC-binding chemokines; IL-8; and regulated on activation, normal T cell expressed and secreted (RANTES) compared with individuals who were negative for the allele, FY*B null . The DARC modulates the bioavailability of proinflammatory chemokines, including RANTES (CCL5) and IL-8 (CXCL8), which are inactivated once bound to RBCs.
Gene expression studies have shown that sickle RBCs, either directly or indirectly, promote endothelial up-regulation of TNF-α and IL-1 genes. Jison and colleagues demonstrated differential expression of 112 genes involved in inflammation, heme metabolism, cell cycle regulation, antioxidant responses, and angiogenesis in peripheral blood mononuclear cells from SCD patients at baseline.
The importance of the vascular endothelium and its participation in the inflammatory response in SCD has become increasingly appreciated and is convincingly related to its activation by inflammatory stimuli and abnormal expression of adhesion molecules. Circulating ECs from individuals with SCA exhibit an activated phenotype with abnormal expression of adhesion molecules, selectins, tissue factor (TF), and up-regulated heme oxygenase-1 (HO-1). Biologic modifiers triggering endothelial activation during sickle vasoocclusive episodes include hypoxia; oxidant molecules; cytokines, in particular TNF-α and IL-1; and thrombin.
Adhesive interactions between sickle RBCs, leukocytes, platelets, and the vascular endothelium cause vasoocclusion, ischemia, and reperfusion injury that result in acute vasoocclusive pain episodes, ACS and, over time, ischemic organ damage. The development of sickle cell mouse models that mimic sickle cell vasoocclusion and ischemia-reperfusion injury in humans has provided critical information about the pathobiology of SCD. Because inflammation plays a significant role in the development of acute clinical manifestations and chronic vascular injury in SCD, pharmacologic strategies aimed at pathways of inflammation may have both therapeutic and preventative value.
This review summarizes the accumulated evidence from clinical and experimental studies that implicate the inflammatory response in the development of vascular injury associated with SCD. Despite the broad impact of inflammation on acute complications and chronic vascular disease in SCD, no directed antiinflammatory therapies for the treatment or prevention of vasoocclusive events currently exist. An integrated approach using a combination of therapeutic agents directed at individual components of the inflammatory response may ultimately be necessary to make a clinical impact on this debilitating disease.
Introduction
SCD is now recognized as a complex disease characterized by acute and chronic inflammation. The incidence of nearly every clinical manifestation of SCD correlates with white blood cell (WBC) count, indicating a role for leukocytes and inflammation in the pathophysiology of SCD. Leukocytosis is common in SCD patients and is manifested by elevation in monocyte and neutrophil counts, accompanied by elevated levels of circulating inflammatory cytokines, including tumor necrosis factor α (TNF-α), interleukin (IL)-1, and IL-8. Elevated levels of these cytokines have been shown to exacerbate sickling in experimental models and correlate with clinical vasoocclusive severity. Leukocytosis is associated with a decreased life expectancy and, when observed in infancy, predicts future disease severity. Clinically, elevated baseline WBC counts have been associated with acute chest syndrome (ACS), decline in lung function, and ischemic stroke in children with sickle cell anemia (SCA). Reports of acute vasoocclusive pain events, ACS multiorgan failure, and death after administration of myeloid colony-stimulating factors (granulocyte colony-stimulating factor or granulocyte-macrophage colony-stimulating factor) suggest that the association of leukocytosis and poor outcome may be causal. The reduced incidence of vasoocclusive pain and ACS episodes in patients on hydroxyurea (HU) is thought due, in part, to the myelosuppressive effect of the drug.
Genetic associations with inflammation also support the involvement of inflammatory pathways in the pathophysiology of SCD and may partly explain the phenotypic heterogeneity of the disease. Associations between variants in the transforming growth factor β1 (TGF-β)/bone morphogenetic protein 6 pathway and pulmonary hypertension, stroke, and osteonecrosis in SCD have been replicated in 4 independent studies.
Studies showing an increased risk of severe disease in SCA patients with elevated levels of TNF-α receptor-1 (TNF-R1) and vascular cell adhesion molecule (VCAM)-1 are supported by findings from a genome-wide association study of a link between disease severity and polymorphisms in VCAM-1 and in ADP-ribosylation factor guanine nucleotide-exchange factor 2 ( ARFGEF2) , a gene involved in TNF-R1 release.
Another recent gene-centric association identified an association between ACS and a variant (rs6141803) located in close proximity to a gene ( COMMD7) that is highly expressed in pulmonary endothelial cells (ECs), interacts with NF-κB signaling, and is differentially expressed when exposed to oxidant heme species.
Individuals with SCA who express the integral red blood cell (RBC) membrane glycoprotein, Duffy antigen receptor for chemokines (DARC), were found to have higher steady-state WBC counts and levels of DARC-binding chemokines; IL-8; and regulated on activation, normal T cell expressed and secreted (RANTES) compared with individuals who were negative for the allele, FY*B null . The DARC modulates the bioavailability of proinflammatory chemokines, including RANTES (CCL5) and IL-8 (CXCL8), which are inactivated once bound to RBCs.
Gene expression studies have shown that sickle RBCs, either directly or indirectly, promote endothelial up-regulation of TNF-α and IL-1 genes. Jison and colleagues demonstrated differential expression of 112 genes involved in inflammation, heme metabolism, cell cycle regulation, antioxidant responses, and angiogenesis in peripheral blood mononuclear cells from SCD patients at baseline.
The importance of the vascular endothelium and its participation in the inflammatory response in SCD has become increasingly appreciated and is convincingly related to its activation by inflammatory stimuli and abnormal expression of adhesion molecules. Circulating ECs from individuals with SCA exhibit an activated phenotype with abnormal expression of adhesion molecules, selectins, tissue factor (TF), and up-regulated heme oxygenase-1 (HO-1). Biologic modifiers triggering endothelial activation during sickle vasoocclusive episodes include hypoxia; oxidant molecules; cytokines, in particular TNF-α and IL-1; and thrombin.
Adhesive interactions between sickle RBCs, leukocytes, platelets, and the vascular endothelium cause vasoocclusion, ischemia, and reperfusion injury that result in acute vasoocclusive pain episodes, ACS and, over time, ischemic organ damage. The development of sickle cell mouse models that mimic sickle cell vasoocclusion and ischemia-reperfusion injury in humans has provided critical information about the pathobiology of SCD. Because inflammation plays a significant role in the development of acute clinical manifestations and chronic vascular injury in SCD, pharmacologic strategies aimed at pathways of inflammation may have both therapeutic and preventative value.
This review summarizes the accumulated evidence from clinical and experimental studies that implicate the inflammatory response in the development of vascular injury associated with SCD. Despite the broad impact of inflammation on acute complications and chronic vascular disease in SCD, no directed antiinflammatory therapies for the treatment or prevention of vasoocclusive events currently exist. An integrated approach using a combination of therapeutic agents directed at individual components of the inflammatory response may ultimately be necessary to make a clinical impact on this debilitating disease.
Overview of sickle cell disease pathophysiology
Vasoocclusion and transient ischemia-reperfusion events underlie the chronic vascular damage that occurs in SCD. Hemoglobin polymerization and depolymerization cause not only RBC sickling and hemolysis but also a cascade of cellular interactions mediated by a host of inflammatory proteins ( Table 1 ).
| Type | Name | Cellular Source | Main Effects |
|---|---|---|---|
| Cytokines | TNF-α, IL-1 | EC, monocytes, neutrophils, mast cells, platelets | EC activation Adhesion molecule expression Coagulation (induces thrombin, PAF) Vascular permeability |
| Acute-phase reactants | IL-6, CRP | EC | Vascular permeability |
| Chemokines | IL-8 | EC, leukocytes, monocytes, mast cells | TNF, IL-1 regulation EC activation Leukocyte activation, chemotaxis |
| PAF | Leukocytes, mast cells | Leukocyte adhesion, chemotaxis | |
| Eicosanoids LTs | PGI2, PGE2 | Leukocytes, mast cells | Vasodilation |
| LTB4 | SRBC adhesion to neutrophils/EC neutrophil chemotaxis, adhesion | ||
| LTE4 | Bronchoconstriction, airway edema | ||
| Phospholipases | sPLA2 | Lipid breakdown (cell membranes), cytokine induction | |
| Vasoactive amines | Histamine | Mast cells, basophils | Vasodilation, permeability |
| Serotonin | Platelets | Platelet aggregation | |
| NO | EC | Vasodilation Antioxidant (–)Adhesion molecule expression (–)Platelet aggregation | |
| ET-1 | EC | Vasoconstriction ROS production | |
| Platelet factors | IL-1, CD40LG | Platelets | EC activation, SRBC adhesion |
| TNFSF14 | EC adhesion molecule expression | ||
| PF-4 | Coagulation; binds heparins on EC surface |
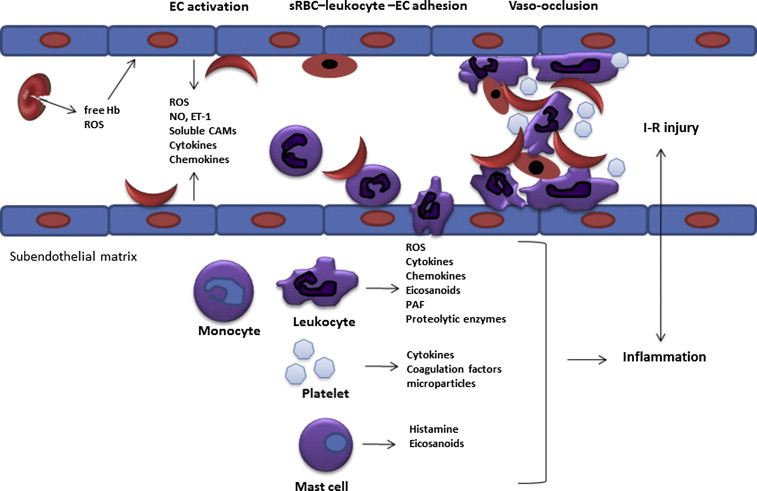
Repeated cycles of sickling cause premature destruction of RBCs with release of hemoglobin and reactive heme iron. These products of hemolysis are a major source of oxidant stress, depleting nitric oxide (NO) through hemoglobin-mediated scavenging, consumption by reactive oxygen species (ROS), and arginase-mediated substrate depletion. Even under ambient conditions, sickle mice develop reperfusion injury with excessive ROS generation in response to hypoxic episodes.
The abnormal adhesion of sickle cells to vascular endothelium involves multiple ligands and receptors and correlates strongly with clinical severity of vasoocclusive pain. Membrane alterations in subpopulations of RBCs expose adhesive receptors and/or phophatidylserine (PS) that promote RBC-endothelial interaction and make them vulnerable to removal or breakdown by enzymes, such as secretory phospholipase A2 (sPLA2), which in turn generates active lipid mediators, such as lysophospholipids and fatty acids, and the release of cytosolic enzymes, including arginase. RBCs show evidence of lipid peroxidation and oxidative damage to structural proteins, and plasma levels of lipid peroxidation products are elevated in SCD patients, indicative of ongoing oxidative stress.
The generation of ROS in sickle RBC is mediated by NADPH oxidase, an enzyme regulated by protein kinase C, Rac GTPase, and intracellular Ca++ signaling and further augmented by TGF-β1 and endothelin-1 (ET-1). Cell-free hemoglobin released by PS-exposing RBC may contribute to NO depletion. Abnormal PS expression on RBC activates the endothelium, permitting exposure of thrombospondin and consequent generation of procoagulants, TF, and thrombin.
The capture of sickle RBCs by adherent leukocytes has been demonstrated both in vitro and in sickle mice and represents a critical step leading to formation of heterocellular aggregates and microvascular obstruction in the process of vasoocclusion. These sickle RBC–leukocyte interactions are provoked by abnormal membrane exposure of PS, intercellular adhesion molecule (ICAM)-4, and autologous immunoglobulins, which can bind leukocyte β2 integrins. Consistent with these results, sickle RBC–leukocyte interactions were abrogated with improvement in blood flow in E-selectin knockout mice, emphasizing the role of E-selectin as a mediator of neutrophil adhesion. Based on these findings, a multistep model for the pathogenesis of sickle cell vasoocclusion has been proposed, highlighting the interaction of adherent leukocytes with sickle RBC and the endothelium ( Fig. 1 ).
Cytoprotective mediators
Nitric Oxide
NO, a potent vasodilator, also mediates inflammation in SCD by countering oxidative stress, down-regulating expression of endothelial adhesion molecules, and inhibiting platelet aggregation.
Patients with SCD exhibit a chronic state of NO deficiency, which is further exacerbated during vasoocclusive pain episodes and ACS as a result of consumption by plasma hemoglobin and ROS (superoxides and xanthine oxidase) generated during ischemia-reperfusion cycles. Endogenously produced inhibitors of NO synthase (NOS), such as asymmetric dimethylarginine, are also increased in SCD patients and may further limit NO availability.
The inhibitory effect of NO on leukocyte adhesion is supported by studies showing that
- 1.
NO donors (eg, nitroprusside) decrease leukocyte adherence induced by inflammation.
- 2.
NOS inhibitors increase recruitment of adherent leukocytes.
- 3.
Depletion of NO by ROS (superoxide) promotes leukocyte adherence.
- 4.
Scavenging of ROS by superoxide dismutase increases NO bioavailability and prevents leukocyte adhesion.
Endothelin-1
ET-1, a potent long-acting vasoconstrictor, counteracts the effects of NO in response to inflammatory stimuli, hypoxia, and shear stress. ET-1 is induced by sickle RBCs in vitro and mediates ROS generation through RBC NADPH oxidase activity. Treatment with an ET-1 receptor antagonist lowered RBC-associated protein disulfide isomerase oxidant activity in sickle mice.
Levels of ET-1 are increased in SCD patients during acute vasoocclusive pain episodes and may remain elevated for weeks thereafter, suggesting that it may be partly responsible for prolongation of vasoocclusive pain symptoms. As the elevation in ET-1 precedes the development of clinical symptoms, blockade of this endogenous vasoconstrictor may ameliorate the disease process. Treatment with HU has also been shown to down-regulate ET-1 gene expression in vitro and is independently associated with a decrease in circulating ET-1 levels in children with SCD.
Adenosine
Adenosine is an endogenous nucleoside that acts to reduce adherence and emigration of leukocytes in inflamed postcapillary venules. The antiinflammatory effects of adenosine have been attributed to the stimulation of the adenosine 2A receptor (A2AR) subtype, expressed on neutrophils, macrophages, platelets, and ECs. Adenosine 2A receptor agonists inhibit oxidative activity and degranulation in neutrophils as well as TNF-α release by monocytes. In preclinical models of SCD, activation of the A2AR in natural killer T cells (NKTs) was shown to reduce NKT-related inflammatory lung injury. In contrast, activation of the adenosine 2B receptor (A2BR) increases RBC 2,3-diphosphoglycerate levels, leading to increased sickling in vitro and to hemolysis, vasoocclusion, and organ failure in sickle mice. Inhibition of the A2BR with an experimental A2BR antagonist, PAB1115, or pegylated adenosine deaminase decreased hypoxia-induced sickling in vitro. These agents produced similar results, with inhibition of sickling, vasoocclusion, and organ damage in sickle mice.
Heme oxygenase-1
Free heme and iron released by hemolyzed RBCs promote inflammatory injury via activation of innate immune responses in macrophages and monocytes. Heme oxygenase and biliverdin reductase signaling detoxify heme and iron and provide catalytic antioxidant, antiproliferative, and antiinflammatory protective signaling. Expression of HO-1 is increased in sickle mice and in SCD patients, in response to heme-induced oxidative stress. By increasing ET-1 expression or administering biliverdin, vasoocclusion and subsequent ischemia-reperfusion injury could be prevented in sickle mice. SCD patients have insufficient HO-1 activity, however, required to handle the excessive heme produced by hemolysis and prevent oxidative stress.
Soluble mediators of inflammation
Histamine, Leukotrienes, and Secretory Phospholipase A2
Histamine
Histamine, a potent inflammatory agent stored in mast cells and basophils, is rapidly released via degranulation in response to proinflammatory stimuli. Under physiologic flow conditions in vitro, histamine can induce the adhesion of sickle RBC to vascular endothelium via H 2 and H 4 receptor–mediated expression of P-selectin. The elevated histamine levels observed in SCD patients at baseline and during morphine administration suggest that the activating effect of histamine on the endothelium may have a significant influence on initiating and propagating pain episodes in SCD. In addition, selective blockade of H 2 and/or H 4 receptors has been shown to prevent sickle RBC adhesion, suggesting that concurrent treatment with histamine antagonists may minimize the adverse histaminic effects produced by opioid analgesics.
Leukotrienes
LTs are inflammatory chemokines released by membrane phospholipids via the 5-lipoxygenase (5-LPO) pathway ( Fig. 2 ). Dihydroxy LT (LTB4) and the cysteinyl LTs (CysLTs)—LTC4, LTD4, and LTE4—induce proinflammatory signaling through activation of specific LT receptors on inflammatory cells and the vessel wall. LTB4 induces adhesion of sickle RBCs and recruitment of neutrophils to the endothelium in sickle mice. Plasma LTB4 levels are markedly elevated in SCA patients at baseline and increase further during vasoocclusive pain episodes and ACS. These neutrophil-derived LTB4 effects on RBC-endothelial adhesion are also supported by prior studies documenting an LTB4-associated increase in the ratio of plasma levels of thromboxane A 2 to prostacyclin and the platelet metabolite, 12-hydroxyeicosatetraenoic acid, as well as up-regulation of endothelial vitronectin receptor expression.
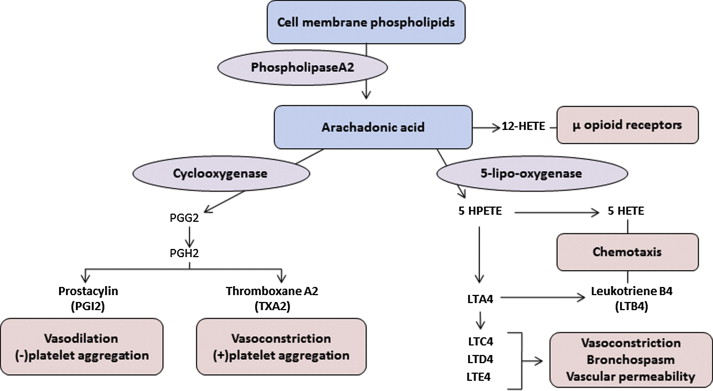
LTB4 is also produced by monocytes to stimulate neutrophil chemotaxis and endothelial adhesion. Monocytes from SCD patients have greater expression of the key catalytic molecules in the LT pathway, 5-LPO and its activating protein (FLAP), than monocytes from healthy controls. This increase in monocyte LT production is mediated by placenta growth factor, indicating another mechanism for the chronic inflammation and increased airway hyperreactivity associated with SCD.
The CysLTs are known for their bronchoconstrictive effects but also potentiate edema, inflammation, and mucus secretion. In children with SCD, urinary LTE4 levels were found elevated and associated with an increased rate of hospitalizations for vasoocclusive pain. A subsequent study confirmed the observed elevation in baseline LTE4 levels and documented further increases in LTE4 levels during acute pain events, suggesting that the proinflammatory and vasoconstrictive effects of LTE4 may be plausibly related to vasoocclusion. Recent studies also suggest that LT and prostaglandins mediate neuropathic pain in SCD. Stimulated and released during vasoocclusive episodes, LTs facilitate the activation of primary afferent nociceptors, resulting in exaggerated pain sensitization.
sPLA2
sPLA2 represents a class of enzymes that hydrolyze phospholipids from cellular membranes and lipoproteins and is implicated in the pathology of a variety of inflammatory conditions, including arthritis, sepsis, and multiorgan failure.
sPLA2 is induced by TNF-α and IL-1 to generate arachidonic acid, lysophophatidic acid, and platelet activating factor (PAF). As such, sPLA2 mediates both proximal and distal effectors of inflammation by catalyzing the production of these compounds and independently inducing ECs and monocytes to release proinflammatory cytokines. In neonates with acute respiratory distress syndrome, sPLA2 levels correlate with alveolar-arterial oxygen gradient, severity of lung injury, and mortality. Styles and colleagues found that baseline levels of sPLA2 are increased in SCD and are further elevated at the onset of ACS, indicating that sPLA2 may play a key role in vascular endothelial damage. Because sPLA2 levels rise 24 to 48 hours before ACS is diagnosed clinically, sequential measurements of sPLA2 can be used to predict the development of ACS in patients hospitalized for vasoocclusive crisis. In addition, C-reactive protein (CRP) values correlate closely with changes in sPLA2 levels in SCD patients and have been used as a surrogate biomarker for sPLA2.
Coagulation Mediators of Inflammation
Early-response cytokines, in particular IL-1β, TNF-α, and IL-6, induce TF expression on ECs and monocytes and simultaneously down-regulate thrombomodulin, endothelial protein C receptors, and fibrinolysis, thus establishing a procoagulant shift in the hemostatic balance. TF expression is increased in pulmonary ECs and circulating monocytes of sickle mice and can be further induced by hypoxia-reoxygenation. Knockout of the TF gene from ECs in sickle mice led to decreased expression of IL-6 but did not have an impact on coagulation, suggesting that endothelial TF is primarily involved in signaling rather than coagulation. A direct mechanism by which coagulation factors promote inflammation is through binding to protease-activated receptors (PARs) on monocytes and ECs, thereby up-regulating proinflammatory cytokines, such as monocyte chemoattractant protein-1, IL-6, and IL-8. Several studies have demonstrated that TF/factor VIIa–dependent activation of PAR-2 promotes inflammation.
Platelet-associated CD40 Ligand
Platelet-associated CD40 ligand (CD40LG), a ligand for the CD40 TNF receptor superfamily protein, has a significant inflammatory and activating effect on ECs. The soluble form of CD40LG has been found elevated and biologically active in individuals with SCA, suggesting a role for platelet-derived mediators in the chronic inflammatory and hypercoagulable state associated with SCD. Elevated CD40LG has been associated with an increased frequency of pain episodes and correlated with leukocyte and platelet counts in patients with SCA.
Platelet-associated TNFSF14
Platelet-associated TNF ligand superfamily 14 (TNFSF14) levels are known to be elevated in SCA patients and induce up-regulation of ICAM-1, VCAM-1, IL-8, and CCL2. Plasma TNFSF14 levels are elevated in SCA patients and correlate with platelet-associated inflammatory markers, CD40LG, IL-8, and ICAM-1. Platelet expression of TNFSF14 is also increased and correlates with platelet activation as measured by P-selectin, suggesting a mechanism by which platelets localize to the vessel wall and release inflammatory mediators. Plasma TNFSF14 has also been associated with elevated tricuspid regurgitant velocity (≥2.5 m/s) in SCA patients and may contribute to the endothelial activation and inflammation associated with pulmonary hypertension in SCA. SCA patients treated with HU had higher levels of plasma TNF receptor superfamily 6B, a soluble receptor that attenuates the effects of TNFSF14, compared with untreated patients, suggesting another beneficial effect conferred by HU.
Cytokines and Chemokines
The interaction of monocytes with the endothelium plays a pivotal role in initiating the inflammatory response by inducing endothelial E-selectin, ICAM, and VCAM and by secreting cytokines, TNF-α and IL-1. These cytokines, together with ROS produced by sickle cell–endothelial interactions, signal the endothelial transcription factor, NF-κB, to produce additional cytokines and chemokines, including IL-8, PAF, and IL-6, all of which contribute to pancellular activation and a second surge of proinflammatory and procoagulant molecules. As discussed previously, many of these inflammatory mediators are chronically elevated in SCD patients, becoming further elevated during acute vasoocclusive pain events and ACS.
A major stimulant of the acute-phase response, IL-6 has been identified as an independent predictor of peripheral vascular disease progression in patients with atherosclerosis. In SCD, IL-6 is liberated at sites of vasoocclusion and stimulates hepatic production of acute-phase reactants, such as CRP, fibrinogen, and haptoglobin. The rise in acute-phase proteins is thought to mitigate tissue damage caused by microvascular ischemia.
Neutrophil activation
In a sickle cell mouse model of vasoocclusion triggered by hemolytic transfusion reaction, Jang and colleagues found that acute vasoocclusive episodes were associated with a striking elevation in neutrophil chemokine CXC motif ligand 1 (CXCL1). Injection of recombinant CXCL1 also induced acute vasoocclusion in sickle mice, as demonstrated by a reduction in microvascular blood flow and leukocyte adhesion to RBC and endothelium. Blockade of CXCL1 receptor, CXCR2, prevented acute occlusion and prolonged survival in these mice. These data suggest that targeted inhibiton of CXCL1 and/or CXCR2 may be a potential therapeutic strategy to prevent acute VOC in SCD patients.
Neutrophil adhesion is enhanced by IL-8 through an increase in cyclic adenosine monophosphate–protein kinase A signaling. Neutrophils isolated from sickle cell patients exhibit increased expression of adhesion molecules compared with neutrophils from healthy control subjects, making them more vulnerable to inflammatory stimuli. Adhesion assays using immobilized fibronectin and TNF-α–stimulated ECs confirm the heightened adhesive properties of neutrophils observed in sickle cell patients.
In addition to stimulating adhesion, IL-8 promotes neutrophil degranulation, oxidative burst, and lipid mediator synthesis. Activated neutrophils thus contribute to oxidative stress by releasing proteolytic enzymes and forming ROS, including superoxide and hydrogen peroxide. Myeloperoxidase (MPO) is also released, binds to ECs, and generates oxidants that scavenge NO and impair endothelial function. Elevated plasma MPO levels have been demonstrated in sickle mice and humans with SCD. In sickle mice, treatment with a novel MPO inhibitor decreased ROS production and lipid peroxidation, with restoration of endothelial- and eNOS-dependent vascular function. Release of cathepsin G and elastase by neutrophils activates both coagulation and platelets. Increased levels of these neutrophil-derived mediators have been associated with acute pain episodes in SCA patients. Platelet-neutrophil aggregates (PNAs) formed in response to inflammatory stimuli are present in patients with septic shock and are elevated in SCD. In sickle mice, PNAs showed greater oxidative activity than activated neutrophils alone. Hypoxia-reoxygenation induced a further increase in PNAs and additional activation of both platelets and neutrophils. Pretreatment with antiplatelet agents, such as clopidogrel or P-selectin antibody, resulted in decreased formation of PNAs, a reduction in neutrophil activation and decreased lung vascular permeability in these mice.
Neutrophil extracellular traps
Activated neutrophils have also been shown to form neutrophil extracellular traps (NETs), a meshwork of nuclear DNA and histones containing granular proteins, such as elastase, cathepsin G, and MPO. NETs formation is mediated by ROS production involving NADPH oxidase and MPO and has been documented in sepsis, malaria, systemic lupus erythematosus, and cystic fibrosis. Aberrant NET formation contributes to tissue damage and predicts multiorgan failure and sepsis. In trauma patients, excessive release of extracellular histones and nucleic acids has been associated with impaired thrombin generation, hyperfibrinolysis, and platelet activation. Using plasma levels of circulating nucleosomes and neutrophil elastase–α 1 -antitrypsin complexes as markers of neutrophil activation and NET formation, Schimmel and colleagues found greater NETs in SCD patients with vasoocclusive pain compared with patients at steady state. The highest nucleosome levels were observed in patients with ACS, paralleling those found in patients with severe sepsis.
Mast cells in inflammation
Mast cells also release inflammatory mediators and proteases, including TNF-α, that contribute to the heightened inflammation observed in SCA. Mast cell activation was associated with increased expression of Fcγ receptor 1 and Toll-like receptor 4 as well as an increase in circulating acute-phase proteins in sickle mice. Mast cell activation also occurs in response to ischemia-reperfusion injury, influencing leukocyte adhesion and transmigration. In sickle mice, pharmacologic inhibition of mast cells with cromolyn or imatinib has been shown to reduce hypoxia-induced systemic and neurogenic inflammation.
In a recent exploratory study using carbon-fiber microelectrode amperometry to monitor the impact of sickle cell–induced inflammation on mast cell function, Manning and colleagues found significantly lower serotonin release in sickle mice compared with control mice. The effect of morphine exposure on mast cell opioid receptors was also demonstrated by an increased release of serotonin in normal mice, indicating the potential ability to compensate for sickle cell–induced changes in mast cell function.
Therapeutic implications
Accumulating data from genomic and preclinical studies in SCD have provided new opportunities to develop therapeutic strategies targeting key genetic and cellular mechanisms in the pathogenesis of SCD. Because antisickling agents only partially ameliorate the vasculopathy of SCD, therapeutic agents targeted at pathways involved in inflammation are attractive candidates for further exploration. Pharmacotherapies currently under investigation include agents that inhibit cellular adhesion and target mediators of inflammation.
Inhibitors of Cellular Adhesion
Intravenous gammaglobulin
Intravenous gammaglobulin is currently under clinical evaluation after 2 studies demonstrated a dose-dependent reduction in RBC leukocyte adhesion, improvement in microcirculatory blood flow, and prevention of vasoocclusion in sickle mice. Although intravenous gammaglobulin showed some beneficial effect in reducing duration of pain in a few patients, its safety profile in SCD still needs to be evaluated. A phase 1/2 study is currently evaluating the effect of a single infusion of intravenous gammaglobulin on the duration of sickle cell pain events ( http://clinicaltrials.gov/ct2/show/NCT01757418 ).
Pan-selectin inhibitor (GMI-1070)
GMI-1070 is pan-selectin inhibitor that prevents adhesion between RBCs and circulating monocytes and neutrophils. Initial studies of this investigational agent in a sickle cell mouse model of vasoocclusion analyzed by intravital microscopy demonstrated that through inhibition of primarily E-selectin and P-selectin and, to a lesser extent, L-selectin, the drug decreased adhesion of sickle RBCs to both neutrophils and ECs and decreased adhesion of neutrophils to endothelium. These effects were associated with improved microcirculatory blood flow and improved survival in these mice. A phase 2 trial of this drug was recently completed, demonstrating a reduction in the duration of pain events, length of hospitalization, and parenteral opioid requirement. With these positive results, a phase 3 trial is planned.
Anti–P-selectin monoclonal antibody (SelG1)
P-selectin blockade by the monoclonal antibody, SelG1, inhibited endothelial adhesion molecule (VCAM-1 and ICAM-1) expression and abrogated hypoxia-induced vasoocclusion in sickle mice. A phase 2, multicenter, randomized controlled study assessing the safety and efficacy of this agent in reducing the frequency of vasoocclusive pain events has recently been initiated ( http://clinicaltrials.gov/ct2/show/NCT01895361 ).
Anti–P-selectin aptamer
Treatment with an anti–P-selectin aptamer almost completely inhibited the adhesion of sickle RBCs and leukocytes in sickle cell mice exposed to hypoxia. Increased microvascular flow velocities and reduced leukocyte rolling flux were also observed in these mice. Anti–P-selectin aptamer may thus be useful as a novel therapeutic agent for SCD and warrants further clinical evaluation.
Platelet ADP receptor antagonist (prasugrel)
Therapies targeting platelet activation and interaction with inflammatory cells may represent another therapeutic avenue to prevent inflammation in SCD. The potential of antiplatelet agents to lessen the incidence and severity of pain in SCD has recently been studied in a randomized phase 2 study of the platelet P2Y12 ADP receptor antagonist, prasugrel, in adult SCD patients. Cellular and soluble markers of platelet activation were decreased and in vivo platelet activation was attenuated in the treated group of patients without serious bleeding complications. In addition to confirming safety, the data suggest the efficacy of prasugrel in reducing the rate of pain in SCD. The safety and efficacy of prasugrel for the reduction of vasoocclusive pain events is now being evaluated in children with SCD in a phase 3 trial ( http://clinicaltrials.gov/ct2/show/NCT01794000 ).
Leukotriene Blockade
Targeting inflammatory mediators involved in arachadonic acid metabolism may be a promising approach for the development of novel therapies for the treatment of SCD. Steroids inhibit production of arachidonic acid, thereby blocking synthesis of LTs and prostaglandins. Ketoprophen is believed to block production of 5-LPO and cyclooxygenase. Aspirin and nonsteroidal antiinflammatory drugs block conversion of cyclooxygenase to prostaglandins and thromboxane A 2 . Additionally, aspirin has been shown to induce biosynthesis of a group of bioactive eicosanoids known as the 15-epi-lipoxins or aspirin-triggered lipoxins. Lipoxins are potent inhibitors of leukocyte chemotaxis, adhesion, and transmigration induced by LTs and other inflammatory mediators, which suggests that they are part of innate protective pathways dampening the host inflammatory response. A safety trial investigating the effect of aspirin prophylaxis on decreasing the incidence of ischemic cerebrovascular disease in children with SCD was terminated early due to poor enrollment.
5-Lipoxygenase inhibitor (zileuton)
Placenta growth factor, an angiogenic cytokine produced by hyperplastic erythroid marrow cells and found elevated in SCD, contributes to activation of monocytes and ECs by inducing a key LT synthetic enzyme, 5-LPO. Zileuton is a specific inhibitor of 5-LPO that decreases LT production, and is Food and Drug Administration–approved for treatment of asthma. Administration of zileuton reduced adhesion of neutrophils and sickle RBCs to rat pulmonary vasculature. Zileuton was also found to increase fetal hemoglobin production in erythroid cells in vitro and may have additive/synergistic effects with HU. A phase I study of zileuton in SCD is now being carried out to establish safety and biologic endpoints ( http://clinicaltrials.gov/ct2/show/NCT01136941 ).
NF-κB Inhibition
Many commonly prescribed antiinflammatory drugs, such as aspirin and glucocorticoids, attenuate activation of transcription factors regulating leukocyte adhesion and or cytokine expression. Nonsteroidal antiinflammatory drugs and glucocorticoids exert therapeutic efficacy by preventing the adhesion and influx of leukocytes. High-dose methylprednisolone decreased the duration of severe pain in children and adolescents with sickle disease, but treatment was associated with rebound pain after discontinuation.
Sickle cell mice treated with the polynitroxyl albumin, an antioxidant that inhibits NF-κB activity, also decreased hepatic and pulmonary endothelial VCAM-1 and ICAM-1 expression, inhibited microvascular leukocyte rolling, and reduced stasis after hypoxia reperfusion. Inhibition of NF-κB activation correlated with reduction in CAM expression.
Additional antiinflammatory drugs that inhibit NF-κB and the up-regulation of adhesion molecules have shown promise in preliminary studies. Transgenic sickle cell mice treated with sulfasalazine showed a reduction in activated circulating ECs, accompanied by a marked decrease in leukocyte adhesion and improved microvascular blood flow disease. In a pilot study, the administration of sulfasalazine to sickle cell patients abrogated the abnormal expression of these adhesion molecules in circulating ECs.
Statins
The 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors (statins) have been shown to improve vascular function, independent of their lipid-lowering properties, by restoring NO bioavailability and suppressing the inflammatory response to endothelial injury. Lovastatin inhibited endothelial TF expression in sickle mice after hypoxia-reoxygenation. Treatment with simvastatin prolonged survival in sickle mice after pneumococcal challenge. In vitro studies of simvastatin in SCD indicate that simvastatin reduces endothelial activation and leukocyte adhesion under basal and stimulated inflammatory conditions. Specifically, simvastatin decreased VCAM-1 and ICAM-1 mRNA levels, inhibited TNF-α–induced activation of NF-κB, and enhanced expression of peroxisome proliferator-activated receptor alpha in cultured ECs.
In a pilot study of 25 adult SCD patients with underlying vascular dysfunction, atorvastatin had no effect on endothelial-independent vasodilation but led to an increase in NOS-dependent blood flow, suggesting improvement in endothelial NOS function. Another pilot study of 28 SCD patients treated with short-term simvastatin documented an increase in plasma NO products and a reduction in plasma levels of IL-6, high-sensitivity CRP, and the soluble adhesion molecules, VCAM-1, ICAM-1, and E-selectin. Collectively, these studies have provided the basis for an extended phase 1/2 study investigating the potential clinical efficacy of simvastatin in decreasing the frequency of vasoocclusive pain events ( http://clinicaltrials.gov/ct2/show/NCT01702246 ).
Adenosine 2A receptor agonist (regadenosan)
Invariant NKTs (iNKTs), a subset of lymphocytes that act to propagate the inflammation associated with ischemia-reperfusion injury, are both increased in number and activated in sickle cell anemia. These cells express high amounts of A2ARs that, when activated, reduced inflammation and lung injury in sickle mice. In a phase I trial of regadenoson, a selective A2AR agonist, expression of NF-κB and iNKT activation associated with vasoocclusive pain events was reduced in patients with SCD. A phase 2 study was recently initiated to assess the efficacy of regadenoson in treating vasoocclusive pain events and ACS in SCD ( http://clinicaltrials.gov/ct2/show/NCT01788631 ).
Although it remains to be seen whether these potential antiinflammatory agents prove useful in clinical management of SCD, the experimental and preliminary clinical findings from investigations provide valuable insights into relevant endothelial signaling events and inflammatory responses that lead to vascular injury in SCD. Agents with multiple mechanisms of action or a combination of agents that target various aspects of the pathophysiology of SCD, such as NO regulation, oxidative injury, inflammation, and cell adhesion, may ultimately prove the most effective therapeutic approach.
Financial Disclosure and Conflict of Interest: The author has nothing to disclose.
Related posts:
 Ischemia-reperfusion Injury in Sickle Cell Anemia
Ischemia-reperfusion Injury in Sickle Cell Anemia
 Gene Therapy for Hemoglobinopathies
Cellular Adhesion and the Endothelium
Hemoglobin S Polymerization and Red Cell Membrane Changes
The Role of Adenosine Signaling in Sickle Cell Therapeutics
Gene Therapy for Hemoglobinopathies
Cellular Adhesion and the Endothelium
Hemoglobin S Polymerization and Red Cell Membrane Changes
The Role of Adenosine Signaling in Sickle Cell Therapeutics
 Alterations of the Arginine Metabolome in Sickle Cell Disease
Alterations of the Arginine Metabolome in Sickle Cell Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


