Louis Pasteur established the germ theory of infectious diseases whilst working near Alès, between 1865 and 1870, following his discovery that the two most prevalent diseases of silkworm were caused by different microbes.1 This groundbreaking discovery apparently settled a long discussion between two conflicting theories. The first, in which diseases were considered to be intrinsic, had long held sway over the second, in which diseases were considered to be extrinsic. It rapidly became clear, with the successive identification of numerous microbes, culminating in Koch’s discovery of Mycobacterium tuberculosis in 1882,2 that this new paradigm accounted for the heavy burden of childhood fever and death that had prevailed throughout human history. About half the children born died of fever before the age of 15, and this death toll could be attributed to microbes.3 This theory did not explain why rare children survived infectious diseases and assumed that healthy children had remained free from infection. However, the edifice on which this theory was constructed collapsed between 1905 and 1915, with the gradual realization that most infected individuals remained asymptomatic, often throughout their lifetime. Asymptomatic individuals were found to harbor latent microbes—nonreplicating microbes in a dormant state— such as M. tuberculosis.4 Even more strikingly, actively replicating microbes were found to cause silent infections in other individuals, such infections being termed “unapparent infections” by Charles Nicolle.5 The question of interindividual variability in the course of infection therefore became, and has since remained, a key question in the fields of infectious disease and immunology, and is, arguably, one of the most important questions in biology and medicine. The problem in itself suggests that there are, after all, some intrinsic determinants of disease. The first explanation put forward followed on naturally from another ground-breaking discovery by Pasteur in 1880 to 1881: the prevention of infectious diseases and the foundations of immunology, with the use of attenuated microbes to vaccinate against fowl cholera and sheep anthrax.6 This led to the hypothesis that related, less virulent microbes or smaller amounts of the same microbes had previously immunized the individuals who remained healthy in the course of infection with a microbe virulent enough to kill other individuals. This powerful idea can be seen as an immunologic or somatic theory of infectious diseases. We now know that this acquired immunity (often referred to as adaptive immunity) emerged twice in the evolution of vertebrates, by convergent evolution, that it is lymphoid, and that it involves both genetic and epigenetic components. However, although this theory was considered plausible in adults, and perhaps in teenagers, especially during secondary infections or reactivation from latency, it was less convincing for primary infections in early childhood, which comprised the majority of cases. In this context, a few human geneticists looking at the problem from the complementary standpoints of clinical and population genetics, including Archibald Garrod and Karl Pearson in particular, collected evidence between 1910 and 1930 for strong, germline, genetic determinism, controlling innate immunity against microbes.7,8 As stated by Garrod, “It is, of necessity, no easy matter to distinguish between immunity which is inborn and that which has been acquired.”7
The development of new vaccines and the discovery of sulfonamides and antibiotics during the 1930s rendered these questions obsolete; it became less pressing to understand a problem that everyone thought would soon be resolved. Paradoxically, antibiotics themselves triggered renewed interest in the question in the early 1950s, when a small group of pediatricians in Europe and America noted that rare children suffered from multiple, recurrent infectious diseases, each of which was treated with antibiotics, lacked a major leukocyte subset or gammaglobulins, and shared this phenotype with relatives, consistent with an inheritable trait.9,10,11,12,13,14 This was the birth of the field of primary immunodeficiency (PID), a term apparently first coined in 197115,16 that remains more frequently used than inborn errors of immunity, which was first used in 1966.16,17 These children had major immunologic abnormalities, such as neutropenia,11,14 alymphocytosis,12,13 or agammaglobulinemia.9,10 Moreover, their infectious and immunologic phenotypes often followed a Mendelian pattern of segregation within their families. It is, however, important to bear in mind that the definition of PID established in the 1950s was based on an artificial, as opposed to natural phenotype, because these children with multiple life-threatening infections would have died during their first episode of infection before the advent of antibiotics. Moreover, although the infectious phenotype of these patients led to their investigation and the pattern of inheritance was suggestive of a Mendelian trait, PIDs were classified and defined solely on the basis of the immunologic phenotype. There had been previous descriptions of inherited disorders associated with infectious diseases, such ataxia-telangiectasia in 1926 and 1941,18,19 Wiskott-Aldrich syndrome in 1937,20,21 and epidermodysplasia verruciformis in 193322 (and its viral etiology in 194623), but these disorders were not recognized as PIDs until 1964,24,25 1959,26,27 and 2002,28 respectively. They were long considered from other angles, in the absence of detectable immunologic abnormalities. Autoimmune polyendocrinopathy syndrome with mucocutaneous candidiasis was probably first described clinically between the 1920s and 1940s, but the autoantibodies involved were not described until the 1970s.29,30 More surprisingly, Chediak-Higashi syndrome (CHS) was first described as an inherited predisposition to infections with detectable leukocyte abnormalities in 1943 to 1954,31,32,33 but was not recognized as a PID until 1962.34
Severe congenital neutropenia (SCN) and CHS were among the first PIDs to be described, but these and other inborn errors of myeloid cells, such as chronic granulomatous disease (CGD), which was first described in 1966, and complement defects, first described in 1963 (hereditary angioneurotic edema due to an absence of C1 esterase inhibitor),35 were not included in the first classifications of PIDs, which were restricted to the lymphoid arm of immunity, in 1968,36 1970,37,38 1971,15 1972,39 1973,40 and 1974.41 Over this period, the fields of immunology and PIDs evolved in parallel, focusing on acquired or adaptive immunity, which is sometimes referred to as adaptive or lymphoid immunity. The lymphoid imprint was so strong that the first report on PIDs even distinguished explicitly between these disorders and myeloid abnormalities. Unsurprisingly, given that these PIDs affected the development or function of B- and/or T-cell immunity, these patients displayed multiple, recurrent infectious diseases that were often opportunistic (ie, not seen in patients with apparently intact immunity). Patients with T- and B-cell PIDs also displayed noninfectious, autoimmune, allergic, and in some cases, cancer phenotypes. Despite the broad infectious phenotypes of the patients with complement and phagocyte disorders identified, the first mention of quantitative and qualitative disorders of phagocytes did not occur until 1974, when it appeared, somewhat amusingly, in a preliminary report,42 but not in the official report43 of the second World Health Organization (WHO) international workshop on PIDs. The weaknesses of this definition and classification of PIDs were apparent to some investigators, including Gatti, who ironically compared this system to the ancient Chinese classification for animals.44 The classification nonetheless evolved, as complement disorders were mentioned in 1976 but phagocyte defects were not,45 and qualitative phagocyte disorders (including CGD and CHS) appeared in the third (1978),46 fourth (1983),47 fifth (1986),48 sixth (1989),49 and seventh (1992)50 WHO classifications, although SCN did not. The full range of PIDs was not covered until the eighth WHO classification in 1999,51 and has since been dealt with in the 10th (2003),52 11th (2004),53 12th (2006),54 13th (2007),55 14th (2009),56 and the most recent (15th57) WHO International Union of Immunological Societies reports. Meanwhile, two prominent reviews published in 1984 and 1995 made no reference to phagocyte defects.58,59,60 Some aspects of the history of the field have been covered in at least two reviews.61,62
The common PID classification and the underlying definition of PIDs proved increasingly inadequate and unable to describe the situation in reality, which extended well beyond phagocytic disorders, from the 1990s onwards. The definition and classification of PIDs have always been engaged in an eternal game of catch-up with the reality of the situation on the ground, and this gap between the conservative words used to describe these conditions and the continual discovery of new PIDs, ever increasing the known spectrum of these diseases, has been maintained.63,64 Phenotypic studies in this field have progressed in at least two ways. First, multiple and diverse new phenotypes have progressively been attributed to PIDs, including, of course, autoimmunity,30 malignancy,65 and allergy,66 but also various other phenotypes, such as autoinflammation,67 angioedema,68 hemophagocytosis,69 and thrombotic microangiopathies.70 The underlying mutations involve multiple circuits affecting both myeloid and lymphoid cells. Second, patients with a single infectious disease, and often with a single infectious episode, have been shown to display PIDs.71 Again, the underlying mutations affect both lymphoid and myeloid cells, but they may also, in some cases, concern nonhematopoietic cells. PIDs were initially associated with multiple, recurrent, and often opportunistic infections with an early onset and fatal outcome. They were familial, recessive traits. Exceptions to these rules gradually emerged, beginning with the description of patients with autosomal recessive (AR) defects in the terminal components of complement (C5 to C9), who are specifically susceptible to Neisseria,72 patients with X-linked recessive (XR) lymphoproliferative syndrome, who are susceptible to Epstein-Barr virus (EBV),73 and patients with AR epidermodysplasia verruciformis, resulting in a selective predisposition to infection with skin-tropic, oncogenic papillomaviruses.74 These studies paved the way for the discovery of new PIDs underlying particular infectious diseases in children who were otherwise healthy and normally resistant to other infectious diseases. Children with mycobacterial diseases were found to carry inborn errors of interferon (IFN)γ immunity.75 Mutations in the toll-like receptor (TLR) and interleukin (IL)-1R pathway are associated with pyogenic bacterial diseases, whereas mutations in the TLR3 pathway are associated with herpes simplex encephalitis.76 Finally, inborn errors of IL-17 immunity underlie chronic mucocutaneous candidiasis.77,78 These discoveries indicated that otherwise healthy children with a single infectious disease can display inborn errors of immunity to primary (in cases of acute disease) or recurrent/latent (for chronic disease) infection.71,79
The definition of PIDs is evolving, thanks largely to the clinical delineation and genetic dissection of new phenotypes. These advances are also leading to changes in the classification of PIDs. PIDs were initially classified into two groups (defects of humoral and cell-mediated immunity), then into four major groups (T, B, complement, and phagocyte disorders). The 2011 WHO classification includes up to 10 (somewhat overlapping) categories of PIDs, despite the contentious omission of certain types of PID.79a However, there is no consensus about the definition and classification of PIDs.16 The classification of PIDs principally on the basis of immunologic phenotypes entails a risk of clinical and genetic overlap and of some disorders being ignored. A classification based on clinical phenotype would be more useful at the patient’s bedside, and a classification based on genotype would be more useful at the research bench. However, the apparent lack of a solution to this problem is not a major concern. Hopefully, the classification will improve with the characterization of more PIDs, the deciphering of their genotypes, the dissection of their molecular, cellular, and immunologic phenotypes. and the description of their clinical phenotypes. Like a jigsaw puzzle, every piece should start to fit into place as we approach the solution. This task may not be as vast as it might initially appear, as there are no more than 25,000 genes for 7 billion individuals reproducing every two or three decades. However, we may never reach the asymptote, as there is no such thing as a distinct disease entity, precisely because of the tremendous germline and somatic genetic variability that makes each disease in each patient unique: there are only patients. There is therefore unlikely to be any satisfactory definition and classification of inborn errors of immunity in the near future. This does not matter so long as rapid progress is made in this field, in terms of awareness, diagnosis, treatment, and above all, in explorations of the genotype and intermediate phenotypes of known and new clinical phenotypes.
Why is it so important for the field of PIDs to thrive? This progress is above all important for the patients. As in other areas of medicine, strategies improving our understanding of pathogenesis constitute the only rational approach to improving the clinical management of patients, in terms of the quality of diagnosis, prognosis, and treatment. The availability of blood samples has made it possible to carry out very careful analyses of the relationships between genotypes, cellular phenotypes, immunologic phenotypes, and clinical phenotypes, as for inborn errors of erythrocytes and platelets, perhaps more thoroughly than in other fields of human genetics and pediatrics. For example, the discovery of new PIDs over the last two decades has made it clear that the 10 conventional warning signs used in PID awareness campaigns are completely out of date and require revision.80 Another clinical lesson learned in the last 50 years is that most PID-causing genes are associated with high levels of clinical heterogeneity. Remarkable examples include mutations in the NEMO gene, the effects of which range from death in utero to mild immunodeficiency in adults, reflecting the severity of the biochemical deficit caused by the morbid alleles,81 and mutations in RAG genes, the impact of which ranges from life-threatening severe combined immunodeficiency in infancy to combined immunodeficiency in adults.57 Several genes have even been found to harbor loss-of-function (LOF) and gain-of-function (GOF) mutations. These genes include WASP, LOF mutations, which underlie Wiskott-Aldrich syndrome (WAS), and GOF mutations, which underlie SCN,82 and STAT1, LOF mutations, which underlie mycobacterial or viral diseases, and GOF mutations, which underlie chronic mucocutaneous candidiasis (CMC).78 Similarly, most, if not all of the known clinical phenotypes are associated with high levels of locus and allelic genetic heterogeneity. For example, several agammaglobulinemia-causing autosomal genes were identified following the discovery of BTK mutations in boys with XR agammaglobulinemia.83 A large proportion of the patients with each PID, particularly for the most recently described conditions, do not carry mutations in known morbid genes. It is therefore highly likely that new genetic etiologies will be discovered in the future. The clinical implications of research in this field actually extend well beyond diagnosis and pathogenesis, as the first cases of successful immunoglobulin (Ig) substitution,9 bone marrow transplantation,84 transfusion-based enzymatic replacement,85 and gene therapy86 all concerned children with PIDs. These children were also among the first to benefit from PEGylated enzymatic replacement87 and treatment with recombinant cytokines.88
Perhaps of greater relevance to this book, this field has had extraordinary immunologic implications. One of the pioneers in this field, Robert Good, often referred to PIDs as “experiments of nature,” reviving a line of investigation that began with Harvey and was followed by Osler, Garrod, McQuarrie, and Burnet, among others.89,90 Indeed, physicians and scientists can learn much by deciphering the enigmas posed by the experiment of nature represented by each patient with a PID. No matter how rare a disorder, it can provide considerable insight into the fundamental laws operating in living organisms. More conventional experiments, designed by humans and carried out in animal models, benefit from being carefully thought out and executed in a controlled manner. However, they suffer from the limitations inherent to their experimental nature. Experimental protocols differ from natural processes in many ways (inbred animals, conditions of infection, microbes). Human genetics provides us with a unique opportunity to define the function of host genes in natural, as opposed to experimental, conditions: in a natural ecosystem governed by natural selection.91,92 Immunity in natura can be defined by the careful dissection of PIDs and by other related approaches, such as epidemiologic and evolutionary genetics. The differences between mice and humans are often discussed, and rightly so, as these two species differ in many ways, despite the similar architecture of their immune systems. Beyond these multiple, and in some cases large, differences between humans and mice, there may be major differences between the processes used to study phenotypes, infectious and otherwise, in the two models. For example, experimental infections in mice generally involve inoculation with microbes that have not coevolved with these rodents, via artificial routes of infection and at high doses. By contrast, most human infections are natural, although some experimental infections, such as those caused by live vaccines, have played an important role in the development of this field. The experimental triggers of autoimmunity in mice are also different from those operating in natural conditions in humans. The genetic dissection of PIDs thus provides us with a unique opportunity to cast new light on the function of human genes in a natural ecosystem. Over the years, these observations have provided invaluable insights sometimes at odds with the mouse model.
It will not be possible to cover the entire field in this chapter. More than 200 inborn errors of immunity have been characterized genetically.57,93 Many other PIDs have been described clinically but have no known genetic etiology. Doctors in this field also know that a large fraction of the patients under their care suffer from exceedingly rare disorders, sometimes apparently limited to a single family, not reported in the medical literature. Moreover, many phenotypes might be due to new PIDs. These phenotypes form a large reservoir for the future dissection of new inborn errors of immunity, as previously illustrated by autoinflammation, hemophagocytosis, angioedema, thrombotic microangiopathies, and isolated infectious diseases.64 Finally, we know that most known PIDs are associated with tremendous locus and allelic heterogeneity. With about 25,000 known genes and only 8000 known inborn errors, including about 300 PIDs, we can predict that 10,000 inborn errors of immunity, defined by a specific causal relationship between a genotype and a phenotype, currently await discovery. An estimate of 1000 to 2000 PID-causing loci is not unrealistic. Even with the only 300 or so known PIDs, it is clear that we cannot cover even a substantial fraction of these diseases here. We will therefore focus our review on seven topics, focusing in more detail on a specific syndrome or disease for each and discussing its genetic basis. We will pay particular attention to the immunologic lessons that can be drawn from these experiments of nature.
INBORN ERRORS OF T-CELL DEVELOPMENT OR FUNCTION
In 1950, Glanzman and Riniker described two infants who presented with severe infections, diarrhea, failure to thrive, and disseminated candidiasis, leading to early death. Severe lymphoid depletion tissue was found at the postmortem examination.94 Similar cases of early death due to severe infection (often with weakly virulent microbes), lymphopenia, and agammaglobulinemia were reported a few years later in Switzerland.95 Both male and female patients were affected by this disease, which was named “Swiss-type agammaglobulinemia”96 to distinguish it from Bruton agammaglobulinemia, which affects principally male patients and is characterized chiefly by recurrent bacterial infections.97 The cases described by Glanzman and Riniker94 and by Hitzig et al.95,96 constitute the first description of humans with severe combined immunodeficiency (SCID), a group of conditions characterized by a lack of autologous T cells and extreme susceptibility to infections caused by a broad range of pathogens, including weakly virulent agents. The genetic heterogeneity of SCID became apparent in the 1960s, with the identification of families in which the disease was inherited as an X-linked trait.98 In the 1970s and 1980s, the immunologic phenotype of SCID was shown to be heterogeneous, through the demonstration that all patients had low levels of autologous T-lymphocytes but that only some of these patients displayed an associated decrease in the numbers of B and/or natural killer (NK) lymphocytes.99 This has led to the identification of various immunological subtypes of SCID: a) with a complete absence of T, B, and NK lymphocytes (T-B- NK- SCID); b) T- B+ NK- SCID; c) T- B+ NK+ SCID; and d) T- B- NK+ SCID. Finally, genetic studies over the last three decades have shown that SCID is also highly heterogeneous in terms of its genetics57,100(Table 48.1; Fig. 48.1).
SCID has a prevalence of 1:50,000 to 1:100,000 live births. The study of SCID has played an essential role in the identification of the key mechanisms governing human T-cell development. In several cases, SCID-causing gene defects were identified in patients before the generation of animal models. Moreover, comparison of humans and mice with mutations in orthologous genes associated with SCID has often revealed differences in the immunologic phenotype in these two species, indicating the existence of speciesspecific differences in the mechanisms governing lymphoid development. Finally, the heterogeneity of mutations in individual SCID-causing genes is associated with variability of the clinical and immunologic phenotype, which may include manifestations other than SCID, such as various forms of immune deficiency and immune dysregulation, particularly in patients with hypomorphic mutations allowing residual development of T- and/or B-lymphocytes.101
Molecular Mechanisms Accounting for Severe Combined Immunodeficiency
Defective thymus organogenesis is associated with a profound impairment of T-cell development. The genetic defects affecting the development of the thymic epithelium are extrahematopoietic in nature, but they may greatly disturb T-cell development, in some cases leading to a clinical and immunologic SCID phenotype. DiGeorge syndrome (DGS) results from defects of the third and fourth branchial pouches and is characterized by impaired thymic development, congenital heart disease, hypoparathyroidism, and facial dysmorphisms. Most patients carry a heterozygous interstitial deletion of chromosome 22q11.102 The molecular mechanism by which haploinsufficiency for the genes of the DGS critical region underlies defective thymic development remains unclear. The severity of the thymic development defect varies considerably between patients. Most patients with DGS have moderate T-cell lymphopenia, but a few have complete thymic aplasia (complete DGS) and no circulating T-lymphocytes.102,103,104,105 Another PID in which thymogenesis is impaired is caused by mutations of the gene encoding FOXN1, a transcription factor required for the development and maturation of the thymus stroma and eccrine glands.106,107 Biallelic FOXN1 mutations cause an AR SCID phenotype in which a lack of circulating T-lymphocytes is associated with alopecia.108,109,110 This disease is the human equivalent of the nude phenotype in mice, for which the underlying gene was identified before the discovery of human patients.111
The proliferation and survival of lymphoid progenitor cells are also essential for the generation of a normal number of mature lymphocytes. Some forms of SCID are associated with high rates of apoptosis. In 1972, while investigating polymorphic enzymes as genetic markers, Giblett discovered that the erythrocytes of two infants with SCID displayed no adenosine deaminase (ADA) activity.112 ADA converts adenosine to inosine (and deoxyadenosine to deoxyinosine).113 In patients with AR ADA deficiency, the accumulation of toxic phosphorylated derivatives of deoxyadenosine causes cell death, resulting in extreme lymphopenia, with an almost total absence of T, B, and NK lymphocytes.112,114,115 A few years after describing ADA deficiency, Giblett discovered that a deficiency of another enzyme involved in purine metabolism, purine nucleoside phosphorylase, was also associated with impaired lymphoid development.116
Defect of signaling (impaired positive selection of CD8+ cells)
MHC class II deficiency
CIITA, RFX5, RFXANK, RFXAP
↓↓ of CD4+ cells
N
N
AR
Defect of positive selection of CD4+ cells
Coronin-1A deficiency
CORO1A
↓↓↓
N
N
AR
Defect of thymocyte egress, impaired T lymphocyte survival
ORAI1 deficiency
ORAI1
N (but defective function)
N
N
AR
Defective calcium flux
Autoimmunity, myopathy
STIM1 deficiency
STIM1
N (but defective function)
N
N
AR
Defective calcium flux
Autoimmunity, myopathy
MAGT1 deficiency
MAGT1
↓↓ of CD4+ cells, reduced proliferation
N
N
XL
Defective magnesium flux
Chronic viral infections, lymphoma
DOCK8 deficiency
DOCK8
↓
↓
↓
AR
Defective activation
Severe atopy, severe cutaneous viral infections
CD40L deficiency
CD40LG
N
N
N
XL
Impaired CD40L-mediated costimulation of B and dendritic cells by activated CD4+ cells
Neutropenia, Cryptosporidium infection, biliary tract disease, neuroectodermal tumors; IgM levels are often elevated
AR: autosomal recessive; CD, cluster of differentiation; DNA, deoxyribonucleic acid; gc, germinal center; Ig, immunoglobulin; IL, interleukin; MHC, major histocompatibility complex;
N, normal; NK, natural killer; PNP, purine nucleoside phosphorylase; RAG, recombinase-activating gene; SCID, severe combined immunodeficiency; TCR, T-cell receptor; XL,
X-linked; XLF, XRCC4-Like factor.
* DiGeorge syndrome is most often associated with 22q11 chromosomal deletion; however, other cytogenetic and genetic abnormalities have been also reported in a minority of cases.
† Hypomorphic mutations in these genes have been associated with Omenn syndrome or other conditions with immune dysregulation.
SCID may also be caused by impaired cytokine signaling by T-cell precursors in the thymus. In 1993, two groups demonstrated that the newly cloned IL2RG gene encoding the γ chain of the IL-2 receptor was mutated in male patients with XR SCID, the most common form of SCID in western countries.117,118 XR SCID is characterized by a T-B+ NK- phenotype, indicating that IL2RG mutations are deleterious to the development of both T- and NK lymphocytes. This was a surprising discovery, because disruption of the Il2 gene in mice had been shown to be associated with immune dysregulation rather than SCID.119 It soon became clear that the IL-2Rγ chain was also common to other cytokine receptors, including those for IL-4, IL-7, IL-9, IL-15, and IL-21,120,121,122,123,124,125 leading to its being renamed the common γ chain, γc. In all these receptors, the γc is coupled to the intracellular kinase JAK3, allowing intracellular signaling.126 These discoveries have helped define the molecular basis of XR SCID and to identify other genetic defects causing SCID in humans. In 1995, two groups established that an AR variant of T- B+ NK- SCID (a phenocopy of XR SCID) was due to mutations of the JAK3 gene.127,128 Furthermore, the observation of severely impaired T-cell development in Il7-/- and Il7r-/- mice129,130 led to the discovery of IL7R mutations in patients with AR T- B+ NK+ SCID.131,132 IL-7 provides important signals for the survival and proliferation of thymocytes and peripheral T cells and regulates the rearrangement of T-cell receptor (TCR) genes,133 thus accounting for the absence of circulating T cells in patients with SCID due to IL2RG,JAK3,and IL7R mutations. Interestingly, B-cell development is unaffected in these forms of SCID, but abolished in Il2rg-/-, Jak3-/-, and Il7r-/- mice.134,135,136,137 These data highlight significant species-specific differences in the molecular mechanisms governing lymphoid development. Circulating NK lymphocytes are absent in patients with XR SCID and with JAK3 deficiency, but not in those with IL-7R deficiency, suggesting that impaired signaling through another γc-dependent cytokine may occur. The cytokine concerned is probably IL-15, because IL-15-mediated signaling is essential for human NK cell development in vitro138 and Il15-/- and Il15r-/- mice lack NK cells in vivo.139,140
SCID can also result from impaired TCR gene rearrangement due to abnormalities in VDJ recombination, a process essential for the expression of both the pre-TCR and the mature TCR.141,142 The lymphoid-specific recombinaseactivating gene (RAG) 1 and RAG2 proteins initiate VDJ recombination by recognizing recombination-specific sequences flanking the variable (V), diversity (D), and joining (J) elements of the TCR and introducing deoxyribonucleic acid (DNA) double-strand breaks.143,144 These breaks are eventually repaired through the ubiquitously expressed nonhomologous end-joining (NHEJ) pathway.145,146 RAG1, RAG2, and the NHEJ pathway are also involved in the rearrangement of Ig genes, which is required for expression of the pre-B-cell receptor (BCR) and the BCR.141,143 Mutations of the RAG1 and RAG2 genes,147 and of the genes encoding Artemis,148 DNA ligase IV,149,150,151,152 and DNA-PKcs153 (all components of the NHEJ pathway) result in T- B- NK+ SCID. Mutations of Cernunnos/XLF, another component of the NHEJ pathway, severely impair, but do not completely abolish T- and B-cell development.154,155 The genes encoding Artemis148 and Cernunnos/XLF154 were cloned from humans before they were cloned from mice, the availability of fibroblastic cell lines from SCID patients being essential to this achievement. NHEJ is involved in general mechanisms of DNA repair, including those occurring in nonlymphoid cells. Thus, patients with defects of this pathway also display an unusually high level of cellular sensitivity to radiation, have a higher than normal risk of malignancy, and may present with neurologic problems.146,156 Finally, hypomorphic mutations of genes involved in V(D) J recombination and NHEJ have been identified in patients with “leaky” forms of SCID, in whom the residual development of T- (and in some cases, B-) lymphocytes was associated with autoimmunity and a high risk of lymphoid malignancies.157,158,159
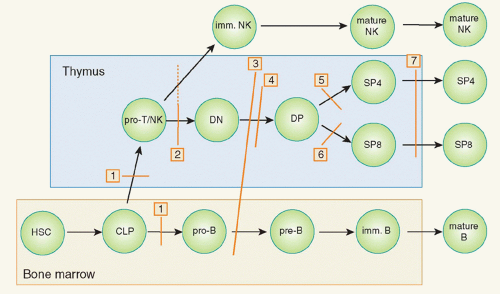
FIG 48.1. Schematic Representation of Developmental Blocks in Combined Immunodeficiencies. Discrete stages in T-, B-, and natural killer (NK)-cell development are shown. Red bars identify stages at which developmental arrests are caused by specific gene defects. Numbers in boxes identify the molecular defects accounting for the developmental blocks. In particular: 1): adenosine deaminase deficiency; 2) defects of cytokine-mediated signaling (IL2RG, IL7R, JAK3 deficiency); 3) defects of V(D)J recombination (RAG1, RAG2, Artemis, LIG4, Cernunnos, and DNA-PKcs deficiency); 4) defects of pre-T-cell receptor signaling (CD3D, CD3E, CD3Z deficiency); 5) defects of positive selection of cluster of differentiation (CD)4+ thymocytes (major histocompatibility complex class II deficiency); 6) defects of positive selection of CD8+ thymocytes (ZAP-70 deficiency); and 7) defects of thymocyte egress from the thymus (Coronin-1A deficiency). The hatched bar indicates that NK-cell development is affected by mutations of IL2RG and JAK3 gene (that interfere with interleukin-15-mediated signaling) but not by IL7R mutations. Not shown is severe combined immunodeficiency due to FOXN1 mutations (that impair thymus organogenesis) and reticular dysgenesis, due to mutations of the AK2 gene, in which the exact stage at which T-cell development is blocked is not known. CLP, common lymphoid progenitor; DN, double negative; DP, double positive; HSC, hematopoietic stem cell; imm., immature; SP4, single positive CD4; SP8, single positive CD8.
Signaling through the pre-TCR is essential to promote the progression from cluster of differentiation (CD)4-CD8- double-negative (DN) thymocytes to CD4+ CD8+ double-positive cells. This signaling is mediated by the CD3 complex, mimicking the requirement of this complex for TCR signaling in mature T cells. Null mutations of the CD3D, CD3E, and CD3Z genes interfere with this process and result in AR SCID.160,161,162,163 By contrast, CD3G mutations are more often associated with a milder phenotype including autoimmunity.164
Other Combined Immune Deficiencies Due to Late Defects in T-Cell Development and Function
The positive selection of CD4+ and CD8+ thymocytes requires the interaction of thymocytes expressing a functional TCR with thymic epithelial cells (and dendritic cells) expressing self-antigens bound to human leukocyte antigen (HLA) class II and class I molecules, respectively.165,166 Several gene defects account for HLA class II deficiency in humans,167,168,169,170 a condition characterized by severe CD4+ lymphocyte depletion and normal CD8+ T-cell development.171 Conversely, mutations of ZAP-70, a tyrosine kinase that binds to the CD3ζ chain and promotes TCR-mediated signaling,172 cause immunodeficiency with a lack of CD8+ cells.173,174,175 The CD4+ lymphocytes of affected patients develop normally but are nonfunctional and fail to proliferate in response to mitogens and antigens. ZAP-70 deficiency in humans results in a phenotype different from that of Zap70-/- mice, which lack both CD4+ and CD8+ lymphocytes.176 The prolonged expression of SYK in human (but not in mouse) double-positive thymocytes seems to compensate for the lack of ZAP-70 protein, allowing the generation of CD4+ T cells.177,178 However, mature CD4+ lymphocytes from ZAP-70-deficient patients have no SYK and are therefore functionally impaired.
The egress of mature thymocytes from the thymus to the periphery requires rearrangement of the actin cytoskeleton. Coronin-1A is involved in this process.179 Accordingly, Coronin-1A deficiency causes the retention of mature thymocytes and a failure to generate peripheral T cells, although impaired survival of newly generated T lymphocytes is probably another very important mechanism of disease.180,181
Finally, mature T cells in the periphery respond to mitogenic signals by releasing calcium from endoplasmic reticulum stores and opening calcium release-activating channels on the cell membrane, allowing calcium influx to occur.182 Mutations of STIM1 (which encodes a sensor of endoplasmic reticulum calcium stores) and of ORAI1 (which encodes calcium release-activating channels) cause a severe immunodeficiency in which T-lymphocyte generation in the thymus is not affected, but the function of peripheral T cells is impaired.181 Both conditions are also characterized by myopathy and the impairment of immune homeostasis, with autoimmunity.183,184
Clinical and Immunologic Features and Treatment
Infants with SCID are susceptible to severe infections from shortly after birth. Infections may be due to bacteria, viruses, or fungi. Infections with weakly virulent microorganisms (eg, Pneumocystis jiroveci, cytomegalovirus) are common and frequently cause interstitial pneumonia and chronic diarrhea, leading to failure to thrive.101,185,186
Lymphopenia is present in 60% to 70% of affected infants.186 T-cell lymphopenia is even more common. In 30% to 50% of cases, a variable number of T-lymphocytes of maternal origin (that have crossed the placenta and not been rejected by the fetus with SCID) are detected.187 Maternal T-lymphocytes may cause alloreactive signs resembling graft-versus-host disease, with infiltration and damage to the liver, the skin, the gut, and the bone marrow.187,188 All patients lack autologous T cells.
Alternatively, less severe (hypomorphic) mutations in SCID-causing genes may be permissive for residual T-cell development, but result in a lower level of TCR diversity in circulating T cells.101 The oligoclonal expansion of these T-lymphocytes is often associated with tissue damage, as in infants with Omenn syndrome.189 This damage resembles the severe inflammatory lesions resulting from the homeostatic proliferation of a few T-cell clonotypes in mice.190 In other patients, the mutations may underlie milder forms of combined immunodeficiency.159,191 However, the severity of the clinical and immunologic phenotype is not determined solely by the nature of the mutation, because different phenotypes (eg, SCID and Omenn syndrome) have been reported in different individuals harboring the same mutation and even in individuals from the same family.192
Lymphocyte and lymphocyte subset counts are the mainstay of SCID diagnosis and may provide useful information about the underlying genetic defect.101 The presence of T cells does not exclude a diagnosis of SCID, as the T cells detected may be of maternal origin. However, the maternal T cells have a very limited in vitro proliferative response to mitogens (eg, phytohemagglutinin).186 Infants with SCID are unable to mount specific antibody responses.186 Genetic diagnosis is guided by the NK- and B-cell phenotype and the mode of inheritance. A SCID screening test for neonates has recently become available. This test is based on the polymerase chain reaction-mediated amplification of TCR excision circles (TRECs) from a dried blood spot collected at birth.193,194 TRECs are a byproduct of TCRα rearrangement that persist in newly developed T-lymphocytes expressing the αβ form of the TCR,195 but are diluted out during the subsequent proliferation of cells in the periphery. TREC quantification therefore provides important information about thymic function.
Infants with SCID require the regular administration of Igs and antimicrobial prophylaxis. When indicated, blood products should be irradiated before transfusion to prevent transfusional graft-versus-host disease, and should be obtained from cytomegalovirus-negative donors to prevent the risk of transmission. The diagnosis and treatment of infection should be prompt and aggressive. However, in the absence of prophylaxis, SCID is inevitably fatal within a few months, and patients do not survive beyond a few years in the absence of transplantation.185 Indeed, hematopoietic stem cell transplantation (HSCT) is the mainstay of SCID treatments, and optimal results (> 90% survival) are obtained if the donor is an HLA-matched sibling.196,197 If no such donor is available, HSCT can be performed with a matched unrelated donor or with a haploidentical parent as the donor. However, if a haploidentical parent acts as the donor, mature T-lymphocytes must be removed from the graft to prevent graft-versus-host disease. Excellent results (> 95% survival) have been reported for SCID patients treated by HSCT before the age of 3.5 months,197 highlighting the importance of neonatal screening for SCID. HSCT results are less satisfactory for combined immunodeficiency with the residual presence of autologous T-lymphocytes, because there is a risk of graft rejection and of treatmentrelated toxicity, due to the need for chemotherapy for ablation of the immune system of the host.196 In particular, the outcome of HSCT is far from good in patients with major histocompatibility complex (MHC) class II deficiency, because the transplantation of hematopoietic stem cells does not correct the lack of MHC class II antigen expression on thymic epithelial cells.198 Enzyme replacement therapy may lead to detoxification and immune reconstitution in infants with ADA deficiency.199,200 Finally, gene therapy has been successfully used in infants with SCID due to ADA deficiency201 or to a γc defect.85,202,203 However, 5 of 20 infants with X-linked SCID treated by gene therapy developed leukemic proliferation due to insertional mutagenesis (ie, insertion of the transgene near an oncogene, mostly LMO2, leading to transcriptional activation of the oncogene).204,205
Thus, studies of patients with SCID have laid the foundations for an understanding of the molecular and cellular mechanisms governing lymphoid development in humans. Identification of the molecular basis of SCID in humans has preceded the generation of gene-targeted mice on a number of occasions. Furthermore, important differences have emerged between the immunologic phenotypes of humans and mice, with mutations in orthologous genes associated with SCID. Finally, the early identification of SCID patients with molecular and immunologic tools has important clinical implications, optimizing survival after HSCT. Despite these advances, the gene responsible for disease remains unidentified in most cases of combined immunodeficiency and a small minority (< 5%) of infants with SCID. The deep sequencing of human exomes and genomes should lead to the identification of still more genes causing T-cell deficiencies in humans.
INBORN ERRORS OF B-CELL DEVELOPMENT OR FUNCTION
Antibody deficiencies constitute the largest group of currently recognized PIDs (Table 48.2).206,207,208 This is partly because these disorders were among the first to be recognized to cause inborn errors in immunity and because they are among the easiest to detect with routine laboratory studies. Antibody deficiencies, regardless of their etiology, are associated with recurrent or persistent infections with encapsulated bacteria, particularly Streptococcus pneumoniae and Haemophilus influenzae. Patients develop infections typical of these organisms, including otitis, sinusitis, and pneumonia, but they are also likely to experience more severe, invasive infections, such as sepsis, meningitis, joint infections, and cellulitis. Other infections and clinical findings are more specific to particular antibody deficiencies.
X-linked Agammaglobulinemia
X-linked agammaglobulinemia (XLA), sometimes called Bruton agammaglobulinemia or congenital agammaglobulinemia, was first described in 1952, when Colonel Bruton reported the case of an 8-year-old boy with recurrent pneumococcal sepsis and no globulin fraction on serum electrophoresis.209 The patient’s clinical course was improved by treatment with exogenous gammaglobulin. Once techniques had been developed for the evaluation of B cells in the peripheral blood, it became clear that patients with XLA had very few or no B cells in the bloodstream.210,211,212,213 This is the most consistent feature in patients with XLA. Although 10% to 15% of patients have higher than expected serum Ig concentrations, all patients have < 2% CD19+ lymphocytes in the peripheral blood (normal range 5% to 20% CD19+ cells). Patients are treated by gammaglobulin replacement and the aggressive use of antibiotics.
In addition to infections caused S. pneumoniae and H. influenzae, patients with XLA are more vulnerable to enteroviral infections, particularly enteroviral encephalitis and vaccine-associated polio.214,215,216,217,218 Mycoplasma and ureaplasma infections are also more common and may cause arthritis, prostatitis, and meningitis, as well as pneumonia.219,220 The incidence of Giardia infection is also high in patients with XLA; some of these patients have protein-losing enteropathy and low serum IgG as the first sign of infection.221 Although significant complications and early death are still sometimes reported for patients with XLA,216 many patients now survive into adulthood.222
In 1993, two groups, using two separate approaches, showed that XLA was due to mutations in a previously unrecognized cytoplasmic tyrosine kinase, now called Bruton tyrosine kinase (BTK).223,224 One group identified the gene on the basis of linkage analysis223; the other isolated a cDNA encoding a tyrosine kinase from B-cell precursors.224 This gene mapped to the XLA critical region on the long arm of the X chromosome, making it an excellent candidate gene.224 BTK is an enzyme specific to hematopoietic cells that is expressed throughout B-cell differentiation.225 It is found in the platelets and myeloid cells, but not in T cells or plasma cells.226 However, the clinical effects of BTK deficiency appear to be limited to the B-cell lineage. Severe neutropenia is seen in some patients with XLA, particularly those who are very young.218,227 However, very low neutrophil counts are also seen in patients with defects specific to the B-cell lineage, such as µ heavy chain deficiency.228 This neutropenia may be due to bone marrow suppression by viruses in the absence of natural antibody.
BTK has an amino-terminal pleckstrin homology domain, a proline-rich region, and an SH3 domain, followed by an SH2 and a carboxy-terminal kinase domain.224 It is activated and phosphorylated within minutes of pre-BCR or BCR activation.229,230 Activated BTK and PLCγ2 then bind to the scaffold protein BLNK, allowing BTK to phosphorylate PLCγ2, resulting in biphasic calcium flux.231
Over 600 different mutations have been identified in the BTK gene,232,233 most of which result in an absence of detectable BTK in monocytes and platelets.234,235,236 Amino acid substitutions, particularly those that do not affect the stability of the protein, tend to result in a milder phenotype characterized by an older age at diagnosis, higher serum Ig concentrations, and the presence of B cells in small, but detectable numbers.237,238,239 However, the genotype/phenotype correlation is not strong.
Mutations of the BTK gene account for 85% of cases of early onset of infection and isolated defects in B-cell development.240 A similar clinical phenotype is seen in patients with mutations in genes encoding components of the pre-BCR and BCR, including the µ heavy chain,228,241,242,243 the surrogate light chain protein λ5,244 the signal transduction molecules Igα and Igβ (also called CD79a and CD79b),242,245,246 and the downstream scaffold protein BLNK.243,247
Bone marrow studies have shown B-cell development to be blocked at the pro-B cell to pre-B cell transition, the stage at which the pre-BCR is first expressed, in patients with mutations affecting BTK, the µ heavy chain, λ5, Igα, Igβ, or BLNK. BTK gene mutations cause a leaky defect in B-cell differentiation. Most patients with XLA have a small number of both pre-B cells in the bone marrow and immature B cells in the bloodstream.248,249,250 As in healthy controls, the number of B cells produced decreases with age,251 and most adults with XLA have less than 0.02% B cells in the blood. Mutations in the other genes cause a more severe block in B-cell development. Null mutations in the µ heavy chain, Igα, and Igβ genes result in a complete absence of CD19+ lymphocytes from the blood (< 0.01%). A patient with a hypomorphic mutation in Igβ and young children with mutations in λ5 or BLNK have been reported to have a small number of B cells (< 0.1% CD19+ cells). By contrast, mice with BTK, λ5, or BLNK defects have been reported to have 50%, 20%, or 10%, respectively, the normal number of B cells in the blood.
TABLE 48.2 Antibody Deficiencies
Disease
Gene Defect
B cells
T cells
Serum Igs
Inheritance
Pathogenesis
Associated Features or Comments
Agammaglobulinemia with absent or very low B cells
Defective initiation of somatic hypermutation and class switch recombination
Lymphadenopathy, increased incidence of autoimmune disease
UNG deficiency
UNG
N
N
↓ IgG and IgA; IgM ↓↓↓
AR
Defective somatic hypermutation and class switch recombination
Lymphoid hyperplasia
AD, autosomal deficient; AID, activation-induced cytidine deaminase; AR, autosomal recessive; BCR, B-cell receptor; CD, cluster of differentiation; CVID, common variable immunodeficiency; Ig, immunoglobulin; N, normal; UNG, uracil-DNA glycosylase; XL, X-linked.
Hyperimmunoglobulin M Syndromes/Class Switch Recombination Defects
Hyper IgM syndrome is characterized by normal or high serum IgM concentrations, with IgG and IgA either present at very low concentrations or undetectable.216,252,253 However, with the identification of the genetic etiologies of the most common forms of hyper-IgM syndrome, it has become clear that not all patients have high IgM concentrations. This has led to the suggestion of “class switch recombination defects” as a more appropriate designation for this group of disorders.253 We will use these terms interchangeably here. Approximately 65% of patients with hyper-IgM syndrome have the XR form of the disease, which is caused by mutations in the gene encoding the tumor necrosis factor (TNF) family member CD40 ligand (CD40L).254,255,256 These patients tend to have more severe illness than patients with XLA.216,252 The median age at diagnosis is 12 months, and patients generally have little or no detectable IgA and IgG in the serum, frequently accompanied by neutropenia and/or opportunistic infections. The numbers of T cells and B cells are usually within normal limits. Infections with encapsulated bacteria, Pneumocystis, cytomegalovirus, parvovirus, Cryptosporidium, and Histoplasma are problematic in patients with CD40L deficiency. Sclerosing cholangitis secondary to Cryptosporidium infection252 and neuroendocrine or hepatobiliary carcinomas257,258 have been reported in a significant number of patients. Treatment consists of gammaglobulin replacement, the aggressive use of antibiotics, granulocyte-colony stimulating factor (G-CSF) in cases of persistent neutropenia and, in some patients, HSCT.259
CD40L (also called CD154, gp39, TRAP, and TNFSF5) is a type II transmembrane protein transiently expressed on the surface of activated T cells260,261,262 and platelets. The binding of CD40L to its cognate receptor on B cells, CD40, induces B-cell activation, short- and long-term B-cell proliferation, and in the presence of cytokines, isotype switching260,262,263,264 through the induction of activation-induced cytosine deaminase (AID) production. By deaminating cytosine residues in VH and switch regions, AID performs the first step in both class switch recombination and somatic hypermutation.265,266 However, CD40 is also expressed on monocytes,267 dendritic cells,268 activated platelets,269 epithelial cells,270 and endothelial cells.271 The stimulation of these target cells via CD40L elicits an inflammatory response, with release of cytokines, including IL-12 in particular.267,272 The failure to elicit this inflammatory response accounts for the viral, fungal, and parasitic diseases seen in patients with CD40L deficiency.
Several other genetic disorders resulting in hyper-IgM syndrome have been reported. A small number of patients with AR defects in CD40 have been described.273 These patients have a clinical phenotype identical to that seen in patients with mutations in CD40L. Approximately 10% to 15% of patients with class switch recombination defects have mutations in AICDA, which encodes the B cell-specific protein AID described previously.266,274,275 Defects in UNG, an enzyme responsible for eliminating the uracil molecules generated by AID activity, also account for a small number of cases.276 Patients with mutations affecting AID or UNG do not have the viral, parasitic, and fungal infections or neutropenia seen in patients with CD40L deficiency, but they are more likely to have lymphadenopathy and autoimmune disease.277
Common Variable Immunodeficiency
Shortly after Bruton described the first case of agammaglobulinemia in a young boy, other authors reported adult patients, of both sexes, with severe hypogammaglobulinemia.278,279,280 Many of these patients appeared to have acquired, rather than congenital immunodeficiency, and both clinical and laboratory findings were highly variable.281,282 This disorder therefore came to be called acquired hypogammaglobulinemia or common variable immunodeficiency (CVID). Hypogammaglobulinemia can be seen in other immunodeficiencies and may result from infections, drug reactions, protein-losing conditions, or cancers, and it is therefore important to rule out these other possibilities before diagnosing CVID.283
Patients of any age may be diagnosed as having CVID, but onset occurs most frequently between the ages of 10 and 40 years.284,285,286,287,288,289 Respiratory tract infections, diarrhea, and autoimmune disorders are the most common findings on presentation. Laboratory analyses show low serum IgG and IgA concentrations, but IgM concentration may be low or within the normal range. B cells are easily detected in the blood of most but not all patients.290 However, most patients lack CD27+ memory B cells.291,292 About 10% of patients have a family history of immunodeficiency or autoimmune disease.286 Patients with CVID or IgA deficiency are more likely to have certain uncommon MHC haplotypes.293,294,295 The precise gene within this locus responsible for susceptibility remains unclear.296 Recent genome-wide association studies have suggested that some copy number variants are more common in CVID.297
In the last 10 years, several genetic disorders resulting in clinical signs consistent with CVID have been reported. A small number of CVID patients with homozygous mutations in ICOS have been identified.298,299 Heterozygous and homozygous mutations affecting TAC1, which belongs to the TNF receptor family (also known as TNFRSF13B), have been reported in 10% of patients with CVID and 1% of healthy controls.300,301,302,303,304 CVID patients with TACI alterations are more likely to have autoimmunity and splenomegaly than other patients with CVID.300,304 Homozygous mutations of the gene encoding another TNF receptor family member, BAFF-R, have been described in a brother and sister in whom immunodeficiency was first recognized after the age of 50 years.290 Homozygous mutations of the genes encoding CD19 or another component of the CD19 complex, CD81, have been reported in fewer than 10 patients.305,306,307,308 A child with a homozygous mutation of the gene encoding CD20 and a low serum IgG concentration has been reported.309 The clinical and laboratory findings for all the individuals with these mutations are highly variable, and it is not clear that any of these genetic alterations are sufficient to cause clinical symptoms when present on their own. In most patients, a combination of genetic and environmental factors is likely to play a role.
INBORN ERRORS OF PHAGOCYTE DEVELOPMENT OR FUNCTION
A number of inborn errors affect the polymorphonuclear or mononuclear phagocytes of the myeloid lineage (Table 48.3). Granulocyte disorders mostly affect neutrophils, whereas mononuclear phagocyte disorders affect monocytes, macrophages, or dendritic cells. Some disorders affect phagocytes and other leukocytes; they may also sometimes affect nonhematopoietic cells. Some of these disorders will be covered elsewhere in this chapter. The disorders mostly or exclusively affecting phagocytes have traditionally been grouped together under the umbrella of “phagocyte disorders.” We will not review all these disorders here, as this group is rapidly expanding. Instead, we will focus on two of these disorders that have played an important role in the history of this field, in which considerable progress has been made in recent decades. These two disorders—severe congenital neutropenia (SCN) and chronic granulomatous disease (CGD), first described by Rolf Kostmann, and CGD, first described by Robert Good— elegantly illustrate the quantitative and qualitative defects of phagocytes. Studies of these disorders have also provided substantial immunologic insight into granulocyte function and the respiratory burst of phagocytes. Many other equally fascinating quantitative defects, such as AR IRF8 deficiency, in which circulating monocytes and dendritic cells do not develop, have been described but will not be discussed here.310 Similarly, we will not consider qualitative disorders here, such as specific granule deficiency, which is characterized by a lack of specific granules, making it impossible to distinguish between neutrophil, basophil, and eosinophil granulocytes, and is caused by AR C/EBP-ε deficiency.311 A list of phagocyte disorders is provided in Table 48.3, under the caveat that the classification of these disorders is imperfect.
SCN is characterized by absolute neutrophil counts of < 500/µl from early infancy, often falling below 200/µl, with normal counts of other leukocytes.312,313,314,315 Patients with SCN have normal counts of basophil and eosinophil granulocytes. Myeloid maturation in the bone marrow is arrested at the promyelocyte or myelocyte stage of development. Differential diagnoses include complex and often syndromic PIDs with neutropenia, such as warts, hypogammaglobulinemia, immunodeficiency, and myelokathexis syndrome, cartilage hair hypoplasia, CHS, dyskeratosis congenita, Fanconi anemia, reticular dysgenesis, and Schwachman-Diamond syndrome. Several other inborn errors of immunity do not affect the myeloid lineage directly but often result in transient or chronic neutropenia, due to infections or the use of particular drugs. Cyclic neutropenia (CN) is characterized by regular oscillations of the number of peripheral neutrophils, with a nadir at about 200/µl and a period of about 21 days. SCN is also known as Kostmann syndrome, and CN is sometimes referred to as cyclic hematopoiesis. Both disorders were long absent from PID classifications, principally because patients with diseases of myeloid cells were seen by hematologists, whereas patients with lymphoid diseases were seen by immunologists. CN is marked by fever, oral ulcers, and bacterial infections during the nadir. The prognosis on G-CSF treatment is excellent316; these patients do not develop myelodysplasia or acute myeloid leukemia (AML).315 The clinical features of SCN are more severe, with mucocutaneous and deep-seated bacterial and fungal infections. The bacterial infections are caused by various Gramnegative and Gram-positive species, including staphylococci in particular. The fungi responsible for infections are equally diverse and include Candida albicans. Without treatment, the outcome in infancy or early childhood is poor. Before the advent of G-CSF treatment, half the patients died from sepsis in the first year of life, the other half dying during early childhood. Myelodysplastic syndromes (MDSs) and AML were rare. Antibacterial and antifungal prophylaxis cannot provide long-term protection. HSCT has been successful in some patients. Spectacular progress was achieved in 1989, with the discovery that recombinant G-CSF could restore normal counts of circulating granulocytes, in at least some patients.87 G-CSF treatment should be tailored to the individual patient. Most SCN patients and almost all CN patients respond to G-CSF treatment, although SCN patients require higher doses. It was subsequently shown that a substantial fraction of SCN patients treated with G-CSF developed MDS or AML, these conditions now constituting a more common cause of death in these patients than sepsis.317,318 No such effect was observed in CN patients. The cumulative incidence of MDS/AML is about 20% after 10 years on G-CSF treatment. Somatic mutations of the G-CSF receptor and monosomy 7 often precede MDS/AML. It is unclear whether G-CSF treatment itself favors these complications or whether treatment reveals natural complications of the underlying disorders. Patients who do not respond to G-CSF, or are undergoing malignant transformation, should be treated with HSCT.319,320,321
Only gold members can continue reading. Log In or Register to continue