58.1
Introduction
Multiple myeloma (MM) is an incurable plasma cell dyscrasia and the second most frequent hematologic malignancy, with an incidence of 5.85/100,000 adults in Western countries and a 5-year survival rate of 48.5% . There were 30,280 new cases of MM diagnosed in the United States in 2017 and 12,590 deaths due to MM . The incidence of MM increases with age, with 70 years being the median age at diagnosis and 2% of cases diagnosed in patients under the age of 40 years. The incidence of MM is slightly higher in males compared to females (1.6:1).
MM evolves from a premalignant condition termed monoclonal gammopathy of undetermined significance (MGUS) that is characterized by the presence of a monoclonal immunoglobulin in the serum at a concentration less than 3 g/dL, with less than 10% clonal plasma cells in the bone marrow, and no end organ damage. Although MGUS affects approximately 2%–3% of people over the age of 50 and increases to 8% of patients over the age of 80, only 1% of MGUS patients progress to symptomatic MM per year . The incidence of MM and MGUS is 2–3 times higher in African-Americans than in Caucasians and parallels the increased incidence of MGUS and MM in West Africans in Ghana . The basis for this higher incidence of MGUS and MM in those of African descent appears to result from a currently unknown genetic predisposition . Recent studies show that African-Americans are more likely to develop specific genetic types of MM than Caucasians .
MM is the most frequent cancer that involves the skeleton, with 90% of patients developing bone lesions over the course of their disease . Bone involvement is responsible for the most devastating consequences of MM, including pathological fractures that can occur in 50%–60% of patients and are already present in 20% of patients at diagnosis. These fractures can cause debilitating bone pain and can increase mortality risk by 20% . MM bone disease (MMBD) can also cause hypercalcemia (15%) and spinal cord compression syndromes (5%) , further impacting the quality of life and survival of patients.
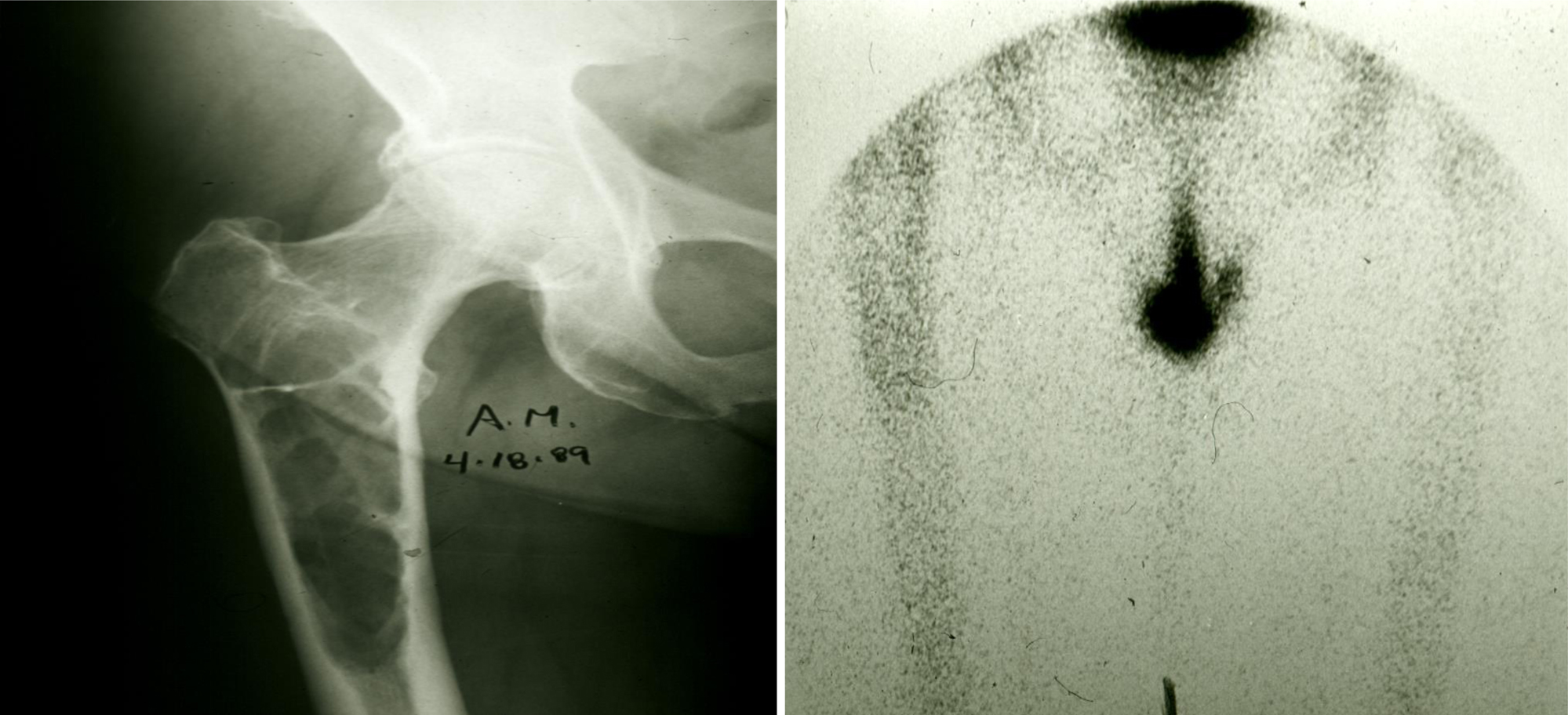
MMBD is characterized by uncoupling of the normal bone remodeling process, with localized increased osteoclastic bone resorption and suppressed osteoblastic bone formation resulting in purely lytic lesions . In the normal bone remodeling process, increased osteoclast (OCL) activity is normally followed by new bone formation at the sites of bone removal. This explains why bone scans, which measure reactive new bone formation, underestimate the extent of bone disease in MM patients ( Fig. 58.1 ). Further, the suppression of bone formation persists even when patients are in complete remission, so that the majority of MM bone lesions do not heal . Recent research has shown that MGUS also impacts the skeleton, with patients having an increased fracture incidence compared to age-matched controls . The mechanisms responsible for the skeletal impact of MGUS and MM on the skeleton and therapeutic approaches to prevent and treat MGUS- and MM-induced bone disease are discussed in the following sections.
58.2
Skeletal effects of monoclonal gammopathy of undetermined significance
As noted earlier, whereas MGUS is uncommon in those aged less than 50 years, the incidence of MGUS increases significantly with aging. Based on prevalence estimates for MGUS and recent US Census Bureau population estimates of 108 million residents aged 50 years or greater, it is likely that MGUS affects approximately 3.5 million US residents. However, because MGUS by definition is not associated with end organ damage, it is most commonly recognized clinically only as an incidental finding during the evaluation of a patient with either signs or symptoms potentially concerning for a lymphoplasmacytic malignancy, or during the evaluation of nonmalignant diseases known to occur more commonly in patients with MGUS (such as osteoporosis), now referred to collectively as monoclonal gammopathies of clinical significance . It is estimated that because only approximately 20% of those with MGUS are likely to have been recognized clinically , roughly 2.8 million persons with MGUS in the United States remain undiagnosed.
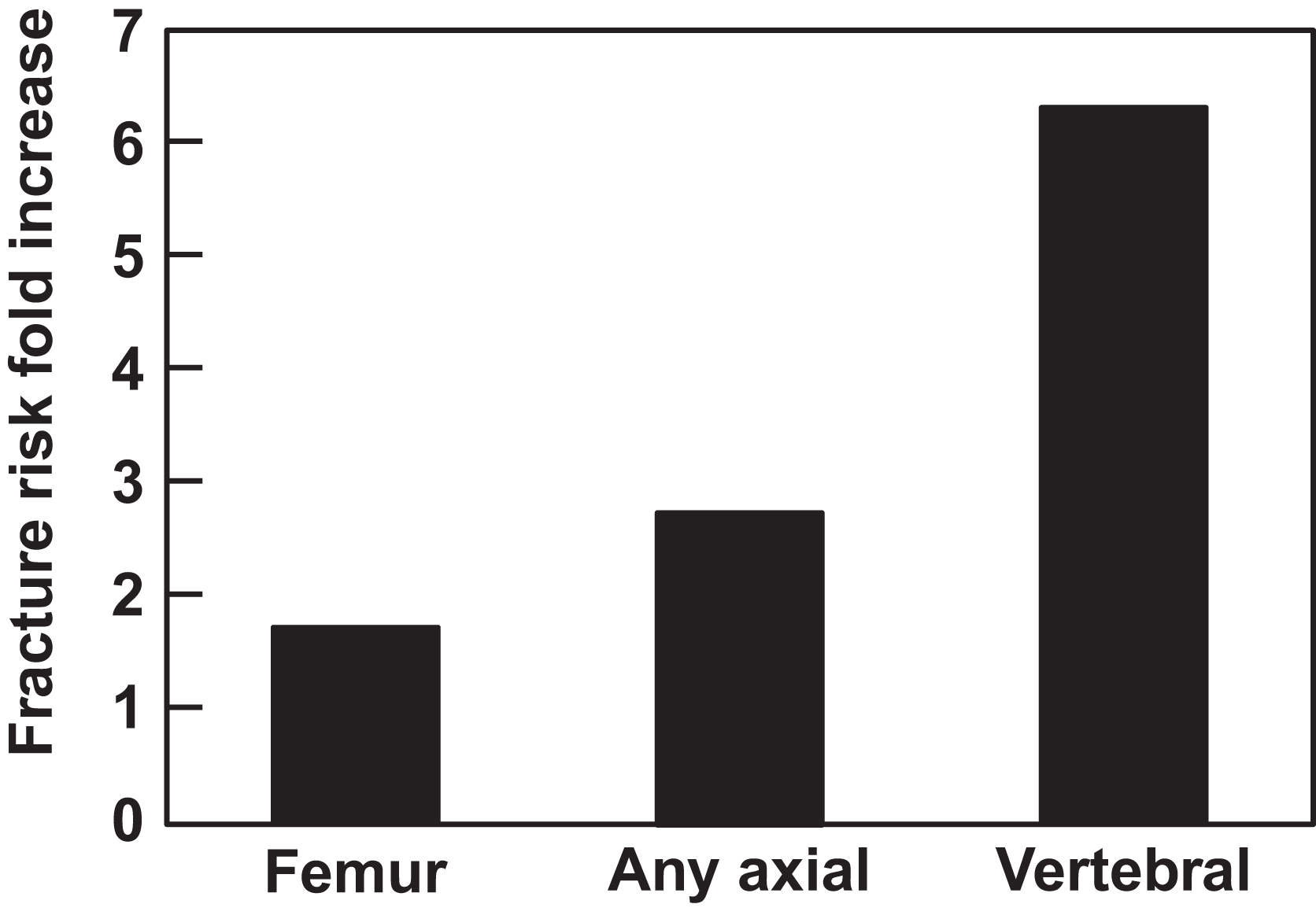
Given that only 1% of most MGUS patients progress to symptomatic MM per year and that the majority of patients have reached at least their seventh decade by the time they are diagnosed with MGUS, most patients are more likely to die from a nonplasma cell-associated condition than due to disease progression. Given this, efforts to limit the risk for MGUS-associated pathologies are important. Of note is that the age-associated increase in risk for developing MGUS occurs in parallel with the inexorable declines in bone mass and increases in fracture risk that result from the normal aging process in both men and women. In patients with MGUS, however, this fracture risk is further increased. As shown in epidemiologic cohort studies performed both in the United States and Sweden, overall fracture rates in MGUS are increased approximately 1.7-fold as compared to matched control subjects without MGUS, with the highest rates (up to 6.3-fold) seen for vertebral fractures ( Fig. 58.2 ). A more recent study, which examined an adult population in Iceland in which all subjects were screened for MGUS, also identified a significantly increased fracture risk of 1.5-fold when only men were considered, although this risk was reduced when women with MGUS were also included in the analysis . In addition, in patients brought to clinical attention due to the presence of osteoporosis either with or without a fracture, the diagnosis of previously unrecognized MGUS is a common finding, as shown in a recent study which reported that 6% of otherwise healthy adults aged >50 years who suffered a hip fracture had concomitant MGUS as compared to an age-matched population where MGUS would be expected to be present in 3% of persons.
Of additional clinical significance is the fact that current National Osteoporosis Foundation guidelines do not recommend screening for MGUS in patients identified by dual-energy X-ray absorptiometry (DXA) as having low bone mass, nor is a diagnosis of MGUS incorporated into the current World Health Organization fracture-risk algorithm used to estimate absolute fracture risk. The prevalence of MGUS in patients without fractures who are diagnosed with osteoporosis by DXA imaging alone is unknown.
As discussed in subsequent sections of this chapter, significant work by multiple groups over the past several decades has identified many of the factors that underlie bone disease in MM and have been shown to be integral for uncoupling of the normal relationship between OCL-mediated bone resorption and osteoblast (OB)–mediated bone formation. Despite MGUS affecting far more persons than does myeloma, however, far less progress has been made in understanding the impact of MGUS on skeletal health.
Clinically, myeloma is characterized both by focal osteolytic lesions as well as systemic bone loss. While focal osteolytic lesions can result in fractures, the systemic bone loss that also occurs predisposes patients with myeloma to develop osteoporotic-type fractures. This duality of fracture risk suggests that local cytokine production from myeloma cells, or cells resident within the bone marrow microenvironment in close juxtaposition to the myeloma cells, results in the development of osteolytic lesions due to local effects on both OCLs and OBs. In contrast, the systemic bone loss that occurs in myeloma is more likely to reflect the effects of circulating factors on bone cell function.
As described, inherent in the clinical definition of MGUS is the absolute absence of end organ damage, including the absence of focal osteolytic lesions. Although osteoporotic-type fractures are increased in MGUS, there has been inconsistent data as to whether bone mass is decreased in MGUS . In part, this confusion reflects limitations of DXA technology. These include the utilization of an areal (two-dimensional) measurement of bone mineral content to approximate volumetric (three-dimensional) bone mineral density (BMD), and the inability to accurately directly assess critical aspects of bone microarchitecture, including disruptions to the trabecular and cortical bone compartments, that are critical for bone quality and maintenance of bone strength.
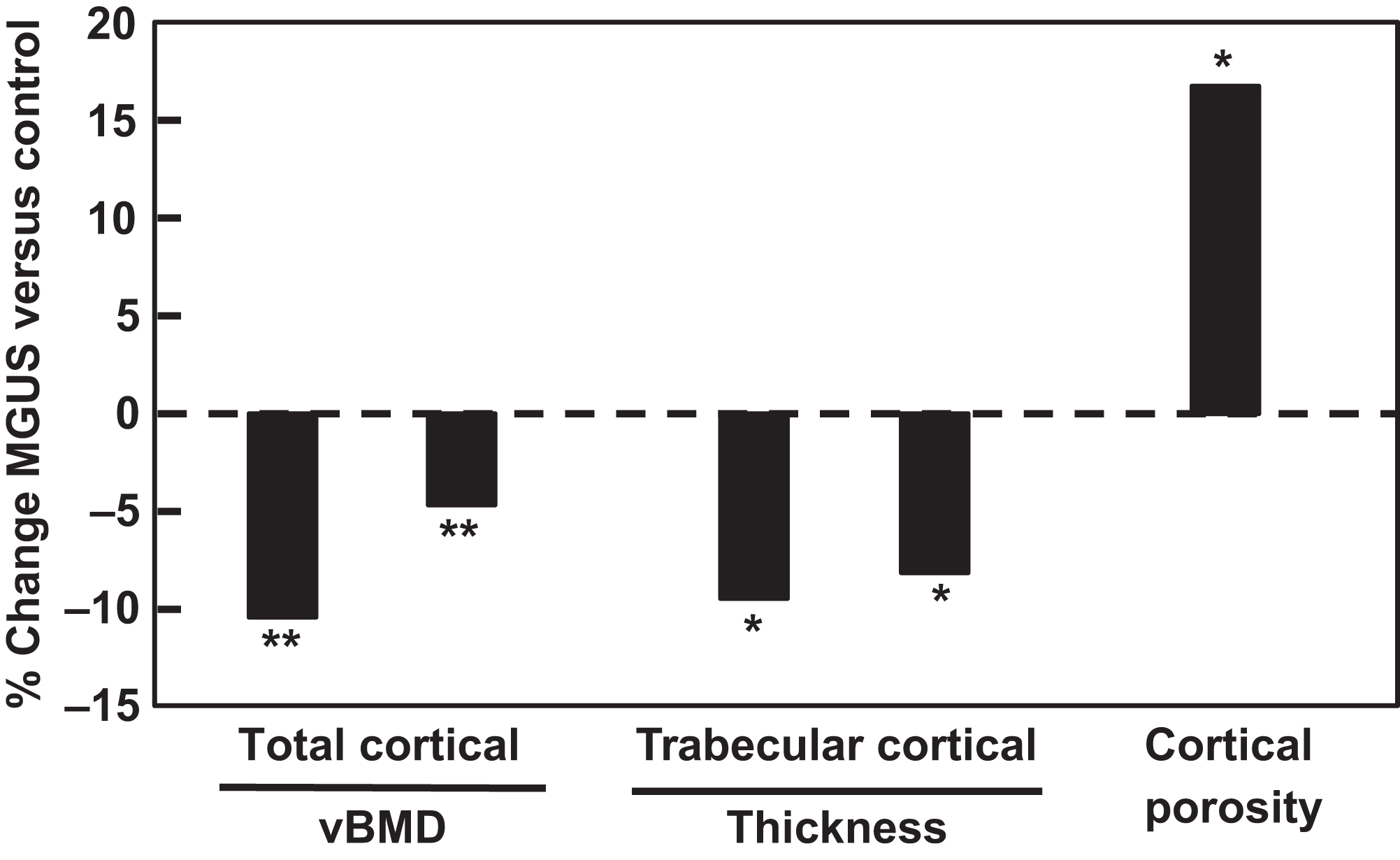
To address this gap in knowledge, the use of advanced imaging techniques, including high-resolution peripheral quantitative computed tomography (HRpQCT), has been employed. In a study which compared standard clinical DXA measurements to HRpQCT measurements in 50 patients with MGUS matched 1:2 to healthy control subjects for age, sex, and body mass index (BMI). Showed that subjects with MGUS showed no differences in femoral neck, spine, whole body, or distal radius areal BMD by DXA. Given that clonal plasma cell levels in MGUS are by definition low (<10% of total bone marrow cells, and usually <5%) and are not evenly distributed throughout the skeleton, it is unlikely that bone marrow changes from the monoclonal plasma cells caused an artifactual increase in DXA-assessed BMD measurements in the MGUS subjects. However, HRpQCT imaging performed at the distal radius showed significantly lower total and cortical volumetric BMD (−10.4% and −4.7%, P <.005, respectively), lower cortical and trabecular thickness (−9.5% and −8.1%, P <.05, respectively), and a substantial increase in cortical porosity (+16.8%, P <.05) in MGUS as compared to control subjects ( Fig. 58.3 ). Collectively, this deterioration of bone microstructure was associated with a significant reduction in biomechanical strength as assessed by microfinite element modeling. Interestingly, MGUS subjects were also found to have a larger radial bone size, which may reflect a compensatory effort to maintain skeletal strength via the apposition of bone to the periosteal surface despite concomitant resorption of bone from the endosteal surface. Similar findings have now been recently reported in a separate cohort of MGUS subjects in a study, which also extended the abovementioned findings to include measurements at the tibia .
The identification and characterization of factors, which impacts bone cell function in myeloma, has been the focus of much effort. Among the factors that have been shown to increase osteoclastic activity are both receptor activator for nuclear factor kappa B ligand (RANKL) and chemokine (C-C) ligand motif 3 (CCL3)/macrophage inflammatory protein (MIP)-1α . In addition, increased circulating levels of factors with the potential to suppress OB activity, including antagonists of the Wnt signaling pathway such as Dickkopf-related protein 1 (DKK1) and the primarily osteocyte-specific protein sclerostin , have been found in patients with MM, with in vitro and in vivo evidence showing their ability to suppress differentiation of mesenchymal stromal cells (MSCs) to OBs . Collectively, these data suggest that the increased osteoporotic-type fractures that occur in patients with myeloma may be at least in part due to the presence of circulating factors.
When assessed by static bone histomorphometry, patients with MGUS display altered bone remodeling, particularly a relative increase in bone resorption . This suggests that alterations in the MSC to OB transition may begin earlier (i.e., when patients have MGUS) rather than later (i.e., once myeloma has been diagnosed), along the monoclonal gammopathy disease spectrum. To date, studies using standard bone labeling methods to assess dynamic histomorphometric indices in patients with MGUS have not been reported. Interestingly, recent work demonstrates that osteocyte dysfunction may be integral to the impairment on bone cell function that occurs in myeloma . Whether disruption in osteocyte function plays a role in MGUS bone disease, however, is unknown.
Similar to myeloma, altered circulating cytokine levels have now also been shown to be present in MGUS. In one study that assessed several factors known to play important roles in myeloma bone disease, circulating levels of the Wnt-pathway inhibitor DKK1 were increased roughly twofold, and levels of the OCL-stimulating factor CCL3/MIP-1α were increased approximately sixfold, in patients with MGUS versus healthy age-, sex-, and BMI-matched control subjects . However, this was not true for all markers, as sclerostin levels were not different between the groups. In aggregate, these data provide early evidence that circulating biochemical factors important for the establishment of bone disease in myeloma are already present at increased levels in MGUS. Further, given data, which show that prior to clinical recognition, more than 50% of patients have had MGUS for at least 10 years and nearly 30% have had MGUS for more than 20 years , it is certainly possible that prolonged increases in the levels of some circulating cytokines common to MGUS and myeloma may lead to subtle systemic alterations in OB and OCL function and ultimately negatively impact whole body skeletal health. In this context, it is noteworthy that while more than 20 additional factors that alter bone cell function have been identified in humans with myeloma, few of these have thus far been formally studied in humans affected by MGUS.
Finally, although higher monoclonal protein levels are positively correlated with progression from MGUS to myeloma, several studies have failed to establish a relationship between monoclonal protein levels and fracture risk . While a recent report did suggest that vertebral fracture risk was substantially higher in MGUS patients with lambda light chain isotype (odds ratio 4.32, P <.01) , this finding was inconsistent with previous reports that found an increased fracture risk with the kappa light chain isotype .
Although there is good evidence that patients with MGUS have an increased fracture risk, our knowledge of the basis for this increased risk remains rudimentary. As an example, there are no studies that have prospectively followed longitudinal changes in BMD in patients with MGUS. Previous small clinical trials have documented that treatment with either oral or IV bisphosphonate is effective for reducing biochemical markers of bone turnover and for increasing BMD assessed by DXA. However, both the routine assessment of skeletal health and the judicious use of bisphosphonates in patients with MGUS remain underutilized . Finally, to date there are no available clinical data regarding the treatment of patients with MGUS with any of the currently available skeletal anabolic agents (teriparatide, abaloparatide, or romosozumab). However, based on their mechanism of action and potential concerns related to the metastatic potential of cancers, which may be tropic to bone (or which grow within the bone marrow—such as myeloma), the use of either teriparatide or abaloparatide may not be considered safe in patients with previously identified MGUS. However, routinely screening for MGUS in patients without a known diagnosis prior to teriparatide or abaloparatide initiation is not considered standard of care and is not required. Whether romosozumab, which increases bone mass via suppression of sclerostin activity, might be a viable option to increase bone mass in patients with MGUS is unknown but likely warrants investigation.
58.3
Skeletal effects of multiple myeloma
58.3.1
Osteoclast-inducing factors
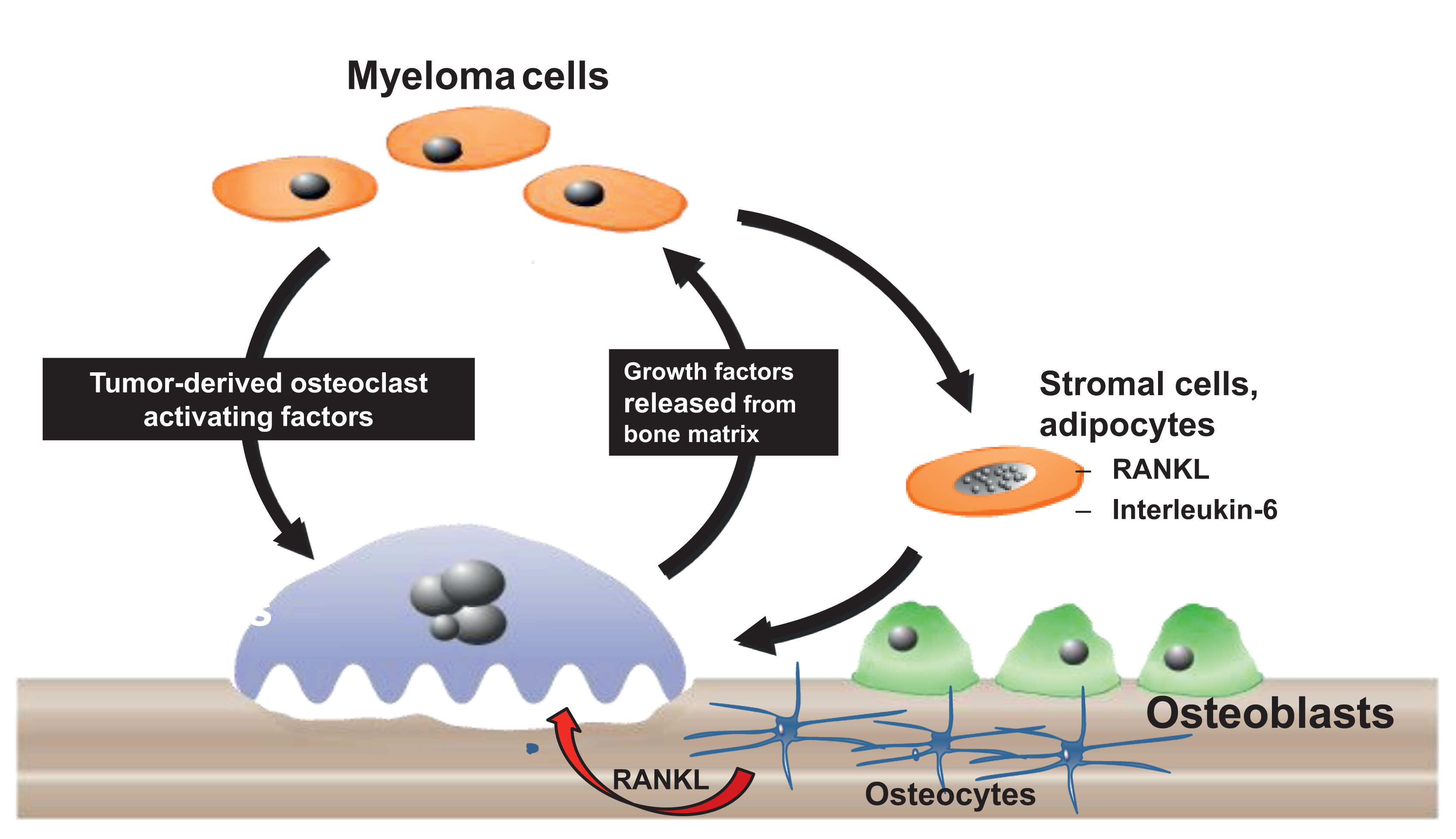
Histological examination of bone biopsies from patients with MMBD showed increased OCL numbers near MM cells. This increase in OCLs is due to local production of OCL-stimulating factors by MM cells or by cells in the bone microenvironment. MM cells secrete multiple factors that can increase OCL formation and activity. These factors include RANKL, (MIP-1α), tumor necrosis factor-α (TNF-α), interleukin (IL)-3, IL-6, parathyroid hormone (PTH)–related protein (PTHrP), and hepatocyte growth factor. These cytokines and chemokines directly or indirectly increase OCL formation and activity . MM cells also increase osteoclastogenesis via interactions with bone marrow stromal cells (BMSCs), osteocytes and T cells in the marrow. For example, adhesive interactions between α4β1 integrin on MM cells and vascular cell adhesion molecule-1 (VCAM-1) on marrow stromal cells promote RANKL, macrophage colony stimulating factor (M-CSF), IL-6, and IL-11 production by stromal cells as well as induce the production of MM-derived MIP-1α, IL-3, and vascular endothelial growth factor (VEGF) . MM cells also physically interact with osteocytes, which also activates bidirectional Notch signaling between osteocytes and MM cells that induce osteocyte apoptosis and increase MM cell growth . Apoptotic osteocytes produce increased amounts of RANKL, sclerostin and DKK1. Sclerostin and DKK1 are Wnt antagonists that inhibit OB differentiation . Importantly, the increased bone destruction induced by MM cells releases growth factors from bone matrix, for example, TGFβ and IGF-1, which increase tumor cell growth . This results in a “vicious cycle” whereby MM cells induce osteoclastic bone resorption that releases growth factors from bone matrix to increase tumor growth and as recently reported, systemic muscle dysfunction ( Fig. 58.4 ).
58.3.1.1
RANK/RANKL/OPG
The RANK/RANKL signaling pathway is a major regulator of both normal and pathological bone remodeling and is a primary driver of OCL activity in MMBD. Although RANKL is expressed by BMSCs, OBs, osteocytes, and activated T lymphocytes in bone , osteocytes are the major source of RANKL during normal bone remodeling and produce 10-fold higher levels of RANKL than OBs . Many cytokines and hormones increase bone resorption, for example,1,25-OH 2 VitD 3 , IL1β and TNF-α, by increasing RANKL expression by OBs and osteocytes . When RANKL binds its receptor RANK that is expressed on the surface of OCL precursors and mature OCLs, multiple signaling pathways are activated, which are required for OCL differentiation, survival, and activity . These pathways include c-Jun N-terminal kinase (JNK), p38, and NF-kB as well as induction of transcription factors c-Fos and NFATc1 . RANKL also binds osteoprotegerin (OPG), a soluble decoy receptor for RANKL produced by BMSCs and osteocytes that compete for RANKL binding to RANK . Multiple studies showed that the RANKL/OPG ratio is a crucial regulator of OCL formation and activity and plays an important role in MMBD . Terpos et al. found that MM patients with an increased serum RANKL/OPG ratio have decreased survival compared to those with lower RANKL/OPG ratio . However, MM cells may produce RANKL or only induce its production by cells in the bone microenvironment. Multiple studies showed that RANKL is expressed by MM cells in bone marrow biopsies and MM cell lines . In contrast, Giuliani et al. found that primary CD138+ MM cells and MM cell lines did not express RANKL, and that increased RANKL/OPG ratios only occurred if human MM cells were cocultured with BMSCs . Regardless, RANKL plays an important role of RANKL in MMBD. Several groups showed in preclinical models of MMBD that blocking RANKL activity with OPG or RANK-Fc decreased both bone destruction and tumor growth in mice . Further, the recent Phase III trial of denosumab in MM patients showed that it increased disease-free survival in addition to decreasing bone destruction .
58.3.1.2
Macrophage inflammatory protein-1α
MM cells and OCLs produce MIP-1α, which is a CC chemokine that increases OCL activity in MM . MIP-1α (CCL3) is a potent OCL-inducing chemokine that can directly stimulate human OCL formation and potentiate the effects of RANKL . Further, MIP-1α enhances myeloma cell adhesion to marrow stromal cells, thereby enhancing marrow stromal cells production of RANKL, TNF-α, and IL-6, as well as VEGF. Elevated MIP-1α gene expression and secretion by MM cells is highly correlated with bone destruction in MM patients and decreased survival . Blocking MIP-1α activity, either with antisense to MIP-1α or treating a mouse model of MM with an antibody to MIP-1α, decreased tumor burden and bone destruction . MIP-1α binds to three different receptors: CCR1, CCR5, and CCR9. CCR1 is the major receptor driving OCL formation in response to MIP-1α, as well as MM cell chemotaxis, growth, and survival induced by MIP-1α . In addition, administration of small-molecule antagonists to CCR1 in models of myeloma blocked tumor growth and bone destruction .
Increased MIP-1 concentrations positively correlate with bone lesions in MM patients, and the osteoclastogenic potential of patient marrow plasma was inhibited by an anti-MIP-1α neutralizing antibody . Further, MIP-1α serum levels are significantly increased in 70% of MM patients with extensive bone disease and are associated with decreased patient survival . MIP-1α can induce OCL formation independent of RANKL , but others reported that MIP-1α did not increase osteoclastogenesis in mice lacking RANK. These results suggest that MIP-1α effects are mediated by inducing RANKL in mice . MIP-1α can also directly impair OB function. Vallet et al. reported that treatment of MM-derived mature OB with MIP-1α prevented OB mineralization activity via downregulation of osteocalcin expression and osterix, and these effects were partially prevented by the small-molecule CCR1 antagonist, MLN3897 in vitro and in vivo . Studies in preclinical models of MMBD showed that blocking CCR1 binding decreases tumor burden and bone destruction in the murine 5TGM1 model of MMBD and partially reverses the inhibitory effects of MIP-1α on OBs. Small-molecule CCR1 inhibitors are under development .
58.3.1.3
Tumor necrosis factor-α
TNF-α simulates osteoclastogenesis by activating a number of signaling pathways, including NF-κB, MAP-kinases, and PI3K/Akt pathways as well as amplifying the effects of RANKL . In contrast, TNF-α inhibits expression of OB differentiation markers, such as Runx2, Osx, type-1 collagen, osteocalcin, and matrix deposition, and induces apoptosis of mature OB . Thus TNF-α increases bone loss and inhibits bone formation. Although TNF-α levels are elevated in the bone marrow microenvironment of MM patients with bone diseases, it is unclear if MM cells produce enough TNF-α to affect bone . TNF-α also increases MM tumor burden by enhancing expression of prosurvival genes, promoting growth and suppressing apoptosis in MM cells by activating several pathways, including the NF-κB pathway . TNF-α is a potent activator of the canonical NF-κB pathway and is itself regulated through this pathway. However, Roy et al. showed that the prosurvival effects of TNF-α in MM cells were due to gain-of-function mutations in the noncanonical NF-κB pathway, which enhanced MM cell resistance to apoptotic stimuli . TNF-α also increases VCAM1expression and RANKL and IL-6 secretion by BMSCs , increasing stromal cell support of OCL activity, MM cell growth, and levels of the angiogenic factors, B-cell activating factor (BAFF) and a proliferation inducing ligand (APRIL), in MM patients .
58.3.1.4
Interleukin-3/activin A
IL-3 is produced by MM cells and T cells in MM patients and is elevated in bone marrow plasma in approximately 70% of MM patients when compared to healthy controls. IL-3 increases OCL-mediated bone resorption and suppresses OB bone formation in MM. Lee et al. showed that IL-3 increases OCL formation by increasing the number of immature OCLs and enhancing the effects of RANKL and MIP-1α . Recently, Silbermann et al. found that IL-3 induces microphage production of activin A from MM patients, a cytokine that can increase OCL formation and inhibit OB differentiation, and that activin A mediated the effects of IL-3 on osteoclastogenesis in vivo . Ehrlich et al. also found that IL-3 blocked OB differentiation through its effects on monocytes/macrophages and inhibited mineralization by mature OB . These data suggest that activin A may mediate IL-3’s inhibition of OB differentiation and regulate the interaction between OCL and OB in MMBD . A soluble activin receptor antagonist sotatercept (ACE-011) is currently in Phase I clinical trial in combination with lenalidomide and dexamethasone for patients with relapsed and refractory MM . Sotatercept can inhibit MM cell growth, improve anemia, and increase bone mass .
58.3.1.5
Semaphorin 4d
MM cells and OCLs produce semaphorin 4d (Sema4D) . Sema4D increases OCL activity and suppresses OB differentiation and motility through binding to its receptor, Plexin-B1 . Both Sema4D and Plexin-B1 levels are increased in MM patients . Recent studies showed that coculture of MM cells with mouse bone increased Sema4D expression in both and that osteocytes were a major source of Sema4D . A Sema4D antibody is in clinical trial for breast cancer bone metastasis .
58.3.1.6
Parathyroid hormone–related protein
PTHrP is functionally similar to PTH, the major regulator of calcium homeostasis . PTHrP acts as an autocrine, paracrine, or intracrine factor for several developmental, physiological, and pathological processes . PTHrP plays a key role in osteolytic metastases induced by different solid tumor types that metastasize to bone . Several studies reported increased serum levels of PTHrP in MM patients with hypercalcemia and osteolytic bone disease compared to MGUS patients or normal donors. These studies also showed that PTHrP is produced by MM cells. It increases tumor cell survival and proliferation and enhances production of osteoclastogenic factors such as RANKL and MCP-1 by MM cells .
58.3.1.7
Interleukin-6
IL-6 is a potent inducer of human OCL formation and is produced by MM cells as well as multiple cell types in the myeloma microenvironment in response to MM cells . IL-6 can directly induce human OCL formation and induce RANKL production and is an antiapoptotic and growth factor for MM cells. Antibodies to IL-6 have been in clinical trials for patients with MMBD. Although a pilot study indicated that blocking IL-6 may decrease the growth of myeloma cells in patients , to date none of these trials have been very positive. These results suggest that targeting IL-6 by itself is not sufficient to inhibit bone destruction and tumor growth in MM.
58.3.1.8
Adhesive interactions
Adhesive interactions between MM cells and BMSCs are involved in MM cell homing, survival, proliferation, and drug resistance. These adhesive interactions activate survival pathways in MM cells, such as NF-κB and p38 MAP-kinase . Activation of these pathways in BMSCs induces the production of osteoclastogenic and angiogenic factors (RANKL, IL-6, and VEGF) that increase OCL formation, angiogenesis and MM survival, and contribute to the bone destructive process .
58.3.2
Osteoblastic suppression in myeloma
Suppression of osteoblastic bone formation plays a critical role in MMBD . MMBD is characterized by an inability of OBs to repair osteolytic lesions even when tumor cells are no longer present in the bone marrow and patients are in long-term remission. Both soluble factors and physical contact between OB progenitors and MM cells suppress OB differentiation and increase OB apoptosis in MMBD . BMSCs from MM patients have an impaired capacity to differentiate into functional OB and express a distinct genomic profile compared with healthy donor BMSCs . It is still unclear if MM cells are required to impair BMSCs from MM patients to differentiate to functional OBs or if it compromised even in the absence of MM cells . The inhibitory effects of MM cells on OB differentiation result from MM cell suppression of the transcription factor Runx2/Cbfal in mesenchymal and osteoprogenitor cells . Giuliani et al. showed that MM patients with osteolytic lesions had a reduced number of Runx2-positive OB compared to patients without MMBD. They found that inhibition of Runx2 in OB progenitors resulted from direct cell–cell contact, via the integrin very late antigen (VLA)-4 on MM cells and VCAM-1 on OB, and that a neutralizing VLA-4 antibody blocked the inhibitory effects of MM cells on Runx2/Cbfal activity . Soluble factors produced by MM cells also contribute to Runx2 suppression (see later).
58.3.2.1
Interleukin-7 and tumor necrosis factor-α
In vivo studies showed that IL-7 suppresses OB differentiation, prevents bone formation, and induces bone loss . Increased IL-7 levels are present in the marrow of MM patients, and neutralizing antibodies to IL-7 partially relieve OB suppression by preventing MM-induced downregulation of Runx2/Cbfal transcriptional activity without affecting its production . Recently, D’Souza et al. reported that IL-7 also induces expression of the transcriptional repressor growth factor independent 1 (Gfi-1), which represses Runx2 gene transcription and potentiates the effects of TNF-α on OB suppression . Using chromatin immunoprecipitation analyses (ChIP), we found that MM cells induce epigenetic histone modifications in OB by converting the Runx2-P1 promoter locus from a poised bivalent state to a repressed state, thereby preventing OB differentiation. Gfi-1 has multiple binding sites at the Runx2 promoter and its direct interaction with Runx2 promoter blocks Runx2 expression. Moreover, Gfi-1 recruits histone modifiers (HDAC1, LSD1, and EZH2) to the Runx2 promoter, which decreases H93K9ac and H3K4me3 and increases H3K27me3 modifications that are essential steps for Runx2 repression. These repressive chromatin changes in Runx2 persist even after removal of MM cells. ChIP analysis also found that BMSCs from MM patients have decreased H93K9ac but showed no significant difference in H3K27me3 when compared with healthy stromal cells. Knockdown of Gfi-1 or selective pharmacological inhibition of HDAC1 and EZH2 activity in pre-OB prevented repression of Runx2 and other OB differentiation markers induced by MM and rescued OB differentiation ( Fig. 58.5 ). These data suggest that treatment of MM patients with clinically available HDAC1 and EZH2 inhibitors might reverse the profound OB suppression associated with MM, possibly allowing the repair of osteolytic lesions .
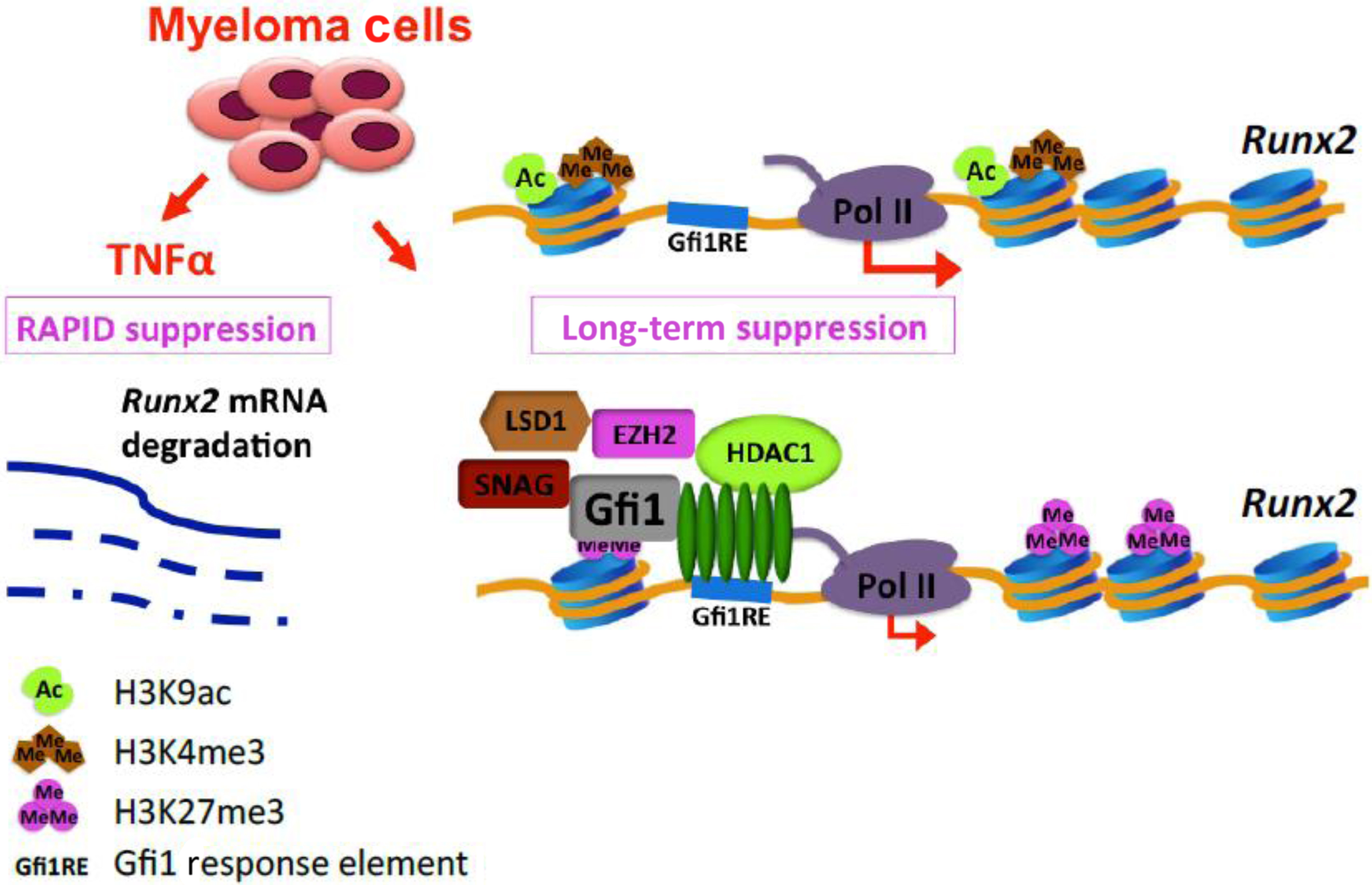
58.3.2.2
Wnt signaling inhibitors
Wnt signaling pathway antagonists also play an important role in OB suppression in MM. Activation of Wnt signaling in OB precursors promotes OB differentiation and survival and increases bone formation . Several groups reported that MM patients have high serum levels of potent Wnt signaling inhibitors, DKK-1 and sclerostin . DKK-1 is produced by MM cells, OBs, and osteocytes and blocks canonical Wnt/β-catenin signaling by preventing Wnt-ligand binding to LRP5/6, thereby downregulating RUNX2 activity to inhibit OB differentiation . DKK-1 also increases RANKL and decreases OPG expression in OBs, which increases osteoclastogenesis and bone destruction . Importantly, Heath et al. found in a preclinical model of MMBD that treatment of MM cells with an anti-DKK-1 neutralizing antibody inhibited MM growth and prevented the development of bone disease . Further, a clinical trial of BHQ880, an anti-DKK-1 antibody, in MM patients treated with melphalan and prednisone, found that BHQ880 used in combination with zoledronic acid may be beneficial in patients with refractory MM and bone disease .
Sclerostin is encoded by the SOST gene and is normally produced by osteocytes. It is a potent inhibitor of canonical Wnt signaling . Expression of several Wnt inhibitors is increased in CD138+ cells from MM patients compared with plasma cells from MGUS patients or normal donors, and their expression is further increased in patients with MMBD compared to MM patients without skeletal lesions . Further, antisclerostin treatment of mice with established MM decreased the number of bone lesions but did not affect MM cell growth . Several studies showed that cancer cells, including breast cancer cells, can also produce sclerostin . Primary MM cells have been reported to express sclerostin , but this remains controversial . Further, other Wnt antagonists are expressed by MM cells , suggesting that Wnt signaling inhibitors, in particular sclerostin, are viable targets for the treatment of MMBD. In addition, treatment of MM patient’s BMSCs with Wnt5a, or overexpression of ROR2, enhanced their osteogenic differentiation, suggesting a potential involvement of the noncanonical Wnt pathway in MMBD .
Osteocyte apoptosis also contributes to MMBD. Giuliani et al. reported that bone biopsies from MM patients had significantly fewer viable osteocytes when compared to control patients, with increased numbers of dead osteocytes and empty lacunae . Osteocytes are the main source of RANKL , and osteocyte apoptosis increases osteocytic RANKL production . We recently found that MM cells increase osteocyte apoptosis via cell–cell contact and/or release of soluble factors by activating the Notch and TNF-α signaling pathways in osteocytes. Osteocyte apoptosis, in turn, increased osteocytic RANKL expression that recruits OCL precursors and osteoclastogenesis. Moreover, physical interaction between MM cells and osteocytes upregulated the expression of Sost/sclerostin in osteocytes that in turn inhibited OB differentiation mediated by Wnt/β-catenin signaling. These results suggest that osteocytes are responsible for the increased sclerostin concentrations in the MM microenvironment . Eda et al. suggested that MM cells also stimulated sclerostin expression in immature OBs via a mechanism that is in part regulated by DKK-1 secreted by MM cells and contributes to impaired OB differentiation in MM . A Phase III trial of romosozumab, a monoclonal antibody that binds sclerostin, showed that romosozumab increased bone formation, decreased bone resorption, and reduced the risk of vertebral fractures in postmenopausal women with osteoporosis . To date, no trials of antisclerostin therapy in MM have been undertaken, but it is an important area of investigation that should be pursued.
58.3.3
Effects of the bone microenvironment on multiple myeloma cells
Recent evidence suggests that marrow adipocytes also play a role in MM. The dynamic interplay between OB, marrow adipocytes, and bone marrow adiposity may create a favorable microenvironment in which MM can engraft and grow and thereby contribute to MMBD. In vitro studies showed that adipocytes support MM cell proliferation and prevent chemotherapy-induced apoptosis by releasing factors such as IL-6, TNF-α, MCP-1, and insulin to increase MM tumor burden and disease progression . The adipokine, adiponectin reduces MM proliferation and induces MM cell death , and reduced circulating levels of adiponectin were reported in MGUS patients that subsequently developed symptomatic MM . Moreover, Fowler et al. recently showed that marrow adiponectin gene expression and adiponectin serum protein levels were significantly reduced in the MM-permissive C57Bl6/KaLwRij mice, compared to the nonpermissive, but closely related C57Bl6/J mice. These authors also found that pharmacological stimulation of adiponectin in tumor-bearing mice decreased MM burden and increased survival of the mice, demonstrating the potential utility of increasing adiponectin for treatment of MMBD .
MM also profoundly modifies the immune function of the bone marrow microenvironment. T cells can modulate bone cell differentiation, survival, and activity . Several studies reported that CD4+ regulatory T cells producing IL-17 are increased in the bone marrow of MM patients . IL-17 can contribute to MM cell survival and is one of the key mediators of lytic bone destruction, which is independent of its effects on tumor cells. Moreover IL-6, which is highly abundant in the MM marrow microenvironment, plays a critical role in the differentiation of CD4+ T cells toward the Th17 lineage via a mechanism that involves TGF-β activation of the retinoic acid–related orphan receptor-γt in a STAT3-dependent manner .
58.3.4
Treatment of multiple myeloma bone disease
Treatment of MMBD is complex and requires the simultaneous management of tumor growth, bone destruction, and suppression of bone formation. Further, MMBD is incurable in most patients and current management utilizes a combination of chemotherapy, localized radiotherapy and surgery.
58.3.5
Bisphosphonates
Bisphosphonate therapy remains the standard care for the management of MMBD . Pamidronate, zoledronic acid, and clodronate are the most commonly used bisphosphonates for treatment of myeloma-induced skeletal-related events (SREs), and reduction of new osteolytic lesions, new pathological fractures, and hypercalcemia . Bisphosphonate treatment also improves bone pain through inhibition of OCL activity. Recently, Hiasa et al. reported that zoledronic acid in combination with selective V-ATPase inhibitor and acid-sensing nociceptor inhibitor significantly reduced MM-induced bone pain in a murine model of MM, by preventing the acidification of the bone microenvironment by proton-secreting OCL . However, bisphosphonate treatments only decrease skeletal-related events by 50% and can decrease renal function. An uncommon but major complication associated with bisphosphonate therapy is osteonecrosis of the jaw (ONJ) .
58.3.6
Denosumab
Denosumab, a highly specific human monoclonal antibody that binds to RANKL and prevents RANKL binding to the RANK receptor on OCLs, is approved by the FDA for the management of bone metastases associated with solid tumors. Data from a randomized, double-blind Phase III study of denosumab versus zoledronic acid in patients with bone metastases or MM showed that denosumab reduced SREs and delayed the appearance of the next SRE as efficiently as zoledronic acid . A recent phase III trial of denosumab versus zoledronate for MMBD was recently completed and showed that patients treated with zoledronic acid had a similar overall survival rate compared to denosumab-treated patients, that both were equally efficacious in preventing SREs and that denosumab increased progression-free survival . Importantly, denosumab could be used safely in patients with impaired renal function and had a similar incidence of ONJ as zoledronate. However, neither denosumab nor zoledronate increases bone formation or repairs lytic lesions in MM patients. Preclinical studies showed that PTH increased bone formation in implanted human bone injected with MM cells . Although, these authors did not find PTH receptors on MM cells, others have reported expression of PTHR1 by MM cells. Thus based on their mechanisms of action and potential to increase the growth of MM cells, it is unlikely that either teriparatide or abaloparatide will be considered safe for use in MM patients.
58.3.7
Proteasome antagonists
Preclinical studies have shown that the proteasome inhibitor, bortezomib and its analogues induce MM cell apoptosis and directly inhibit OCL differentiation and bone resorption, increase osteoblastic bone formation and prevent osteocyte apoptosis induced by MM cells . Retrospective analyses of multiple clinical trials showed that bortezomib and its derivatives increased alkaline phosphatase levels and bone formation markers in responding patients with relapsed MM compared to patients treated with dexamethasone . Although the exact mechanism of OB stimulation by bortezomib in MM patients has not been fully determined, recent studies reported that serum levels of DKK-1, markers of bone resorption and RANKL, were significantly decreased in patients that responded to bortezomib treatment . Several studies demonstrated that patients treated with bortezomib showed signs of bone sclerosis around the lytic lesion, suggesting an initial process of bone healing that was not observed in patients treated only with melphalan and prednisone . A histomorphometric study conducted by Giuliani et al. on bone marrow biopsies of MM patients showed a significant increase in the number of osteoblastic cells/mm 2 of bone tissue and Runx2-positive osteoblastic cells in MM patients responding to the bortezomib treatment . A recent report from Mohan et al. found mineral deposition in large pelvic lesions occurs in a significant proportion of MM patients (43%) treated with very intensive chemotherapy regimen for their myeloma . These results suggest that early eradication of MM cells in patients may allow mineralization of lytic lesions in some MM patients and potentially repair bone lesions.
58.4
Summary
The dramatic bone loss, severe bone pain, and pathological fractures that markedly decrease the quality of life of MM patients result from excessive osteoclastic bone resorption and persistent suppression of osteoblastic bone formation. Studies in patients with MGUS show that bone disease is already present in patients prior to development of MM even though lytic lesions are not detectable in these patients. The lytic lesions that characterize MMBD usually do not heal frequently even when the patients are in complete and prolonged remission, suggesting that bone repair is severely compromised, and may not occur at previous sites of bone destruction. Recent evidence suggests that intensive chemotherapy that reduces tumor burden rapidly may inhibit the progression of MMBD. A clear understanding of the multiplicity of cellular and molecular mechanisms that regulate the interplay between bone marrow cells and MM cells, and the mechanisms responsible for osteoblastic suppression are needed if we are to develop new anabolic agents that in combination with the existing treatments can prevent bone loss, potentially repair, and restore bone damaged by MMBD.
Acknowledgments
This work was supported by grants from VA Merit Review Grant 5101CX000623-05 and NIH 5R01AR057308-09 (G.D.R.).
Conflicts of interest
G.D.R. is a consultant to Amgen. M.T.D. has no conflict of interests regarding the publication of this paper.
References
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


