Drug
Drug specificity
Cut-offs used in trialsa
Unless stated, these represent % tumour cells stained: cell membrane staining
PD-L1 immunohistochemistry primary antibody clone
Complementary or companion diagnostic test
Staining platform required by assay
Nivolumab
Anti-PD-1
≥1%, ≥5%, ≥10%, ≥50%
28-8
Complementary
Dakob link 4800
Pembrolizumab
Anti-PD-1
≥1%, ≥50%
22C3
Companion
Dakob link 4800
Atezolizumab
Anti-PD-L1
TC: ≥1%, ≥5%, ≥50%
IC: ≥1%, ≥5%, ≥10%c
SP142
Complementary
Ventanad benchmark or ultra
Durvalumab
Anti-PD-L1
≥25%
SP263
Not known
Ventanad benchmark or ultra
Avelumab
Anti-PD-L1
≥1%, ≥50%, ≥80%e
73-10
Not known
Dakob, specifics not known
PD-L1 Immunohistochemistry as a Biomarker
PD-L1 IHC and its use as a biomarker for selecting patients for anti-PD-1 or anti-PD-L1 drugs has been a controversial issue [23, 67–69]. This is far from being a perfect biomarker, but no biomarker is perfect. PD-L1 IHC represents a biological continuum of protein expression in different tumours, from nil, through very low, through moderate, to very high numbers of tumour cells expressing the protein. There is evidence or inference from many trials, of a relationship between the amount of PD-L1 in a tumour and the likelihood of response. It is difficult to be certain, but this relationship may not be linear; a certain amount of PD-L1 may be needed, in order to be predictive, but that level is not known. All clinical trials using the biomarker to select a group of patients enriched for chance of response have used a threshold or cut-off level somewhere along the continuum of tumour proportion score. This threshold has been set at 1, 5, 10, 25, 50 or 80% in various trials with various drugs. For atezolizumab , the IHC assay validated in trials also considered the percentage area of tumour infiltrated by PD-L1-expressing immune cells at thresholds of 1, 5 and 10% as an alternative to tumour cell expression, in order to qualify for treatment [65]. These thresholds or cut-offs try to deal with the biological continuum of PD-L1 expression which is not a clear ‘present vs absent’ binary situation (Fig. 20.1). This is quite unlike the other, well-known, addictive oncogene biomarkers such as EGFR mutation or ALK rearrangement, which do represent a clearer selection of patients who will or will not benefit from treatment, biomarkers with a much better performance in terms of predictive power. The use of a cut-off to create the group selected for therapy does not select patients with the same probability of response. The lower the cut point used, the more heterogeneous this responsiveness will be in the treatment group. There is evidence that the average, ‘class’ effect for treatment benefit is largely driven by the high PD-L1 expressing patients in the group. With lower thresholds, there is a risk that patients who have low levels of PD-L1 expression, just above the threshold, will have a significantly less-than-average chance of responding to the therapy. PD-L1 expression can be very heterogeneous in NSCLC, so there is a definite probability of sampling error, in that biopsy samples may not reflect the overall expression in the patient’s disease burden. This error risk is more likely at lower levels of expression. It does mean that some patients will be inappropriately allocated to the wrong side of any threshold being considered. This is probably a major contributing factor to why response rates in groups above threshold are lower than they might otherwise be and why we do see patients who benefit, even if their PD-L1 assessment was considered below threshold or even completely negative. Fortunately, for any given threshold or cut point, most patients’ samples appear clearly above or below the mark, but for those cases close to the cut point, great care needs to be taken to ensure as accurate and consistent an assessment as possible.
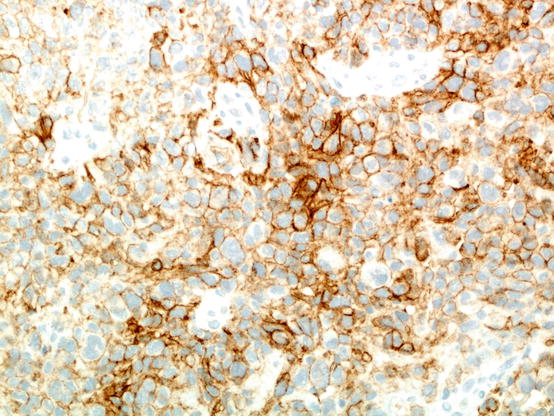
Fig. 20.1
An example of poorly differentiated non-small cell carcinoma stained using the Dako 22C3 anti-PD-L1 immunohistochemistry assay. In this particular field, a majority of tumour cells express PD-L1 on their cell membrane; if this is true for the entire sample assessed, the tumour proportion score would be over 50%
There are currently five drugs targeting the PD-1 axis, either approved or in advanced stages of study, each with their own validated PD-L1 IHC assay (Table 20.1). As multiple drugs get approved, and used in various indications in NSCLC, the pathology community is faced with considerable challenges [23, 67–69]. Each drug-associated assay is different, and there are stringent equipment requirements for each [71]. It would be impractical for most pathology laboratories to offer all five tests, and it may well be impossible, and undesirable, to test the patient’s tumour with all five assays. Instead, can we use any one of the trial-validated assays to stain the sample, but then read the sample according to any number of thresholds that might be required by the oncologist for the patient? Several studies have been undertaken to compare the technical performance in NSCLC samples stained with various trial-validated assays [77–82]. Some of these studies also included laboratory developed tests (LDTs) using a range of anti-PD-L1 IHC clones, some using the same clones as in the trials, and some using clones that have never been used to select any patient in a clinical trial. The findings from these studies may be summarised as follows:
The trail-validated commercially produced assays using the 28-8, 22C3 and SP263 clones show technical equivalence.
When slides stained with these assays were read according to cut points other than the ones used with their associated drug, the concordance for allocating the cases on the same side of the cut points was anywhere between 81 and 95%.
The SP142 assay seems to consistently stain fewer tumour cells compared to the other three assays assessed, leading to lower tumour proportion scores.
All four assays behave similarly in their staining of immune cells.
Some of the LDTs used in the comparisons did match the performance of the comparable trial-validated assays. In one study, however, where several LDTs were developed and compared with the validated assays, about half of these LDTs failed to show adequate concordant staining, even when using the primary IHC clones used in the validated assays [82].
The question for the oncology community is whether this apparent loss of staining performance when using an alternative assay is acceptable. If laboratories choose to use an LDT, there should be a very extensive and rigorous process of validation, in order to ensure that patients are not compromised by being allocated to the wrong treatment group. It is not clear what this validation process should be. Although the College of American Pathologists has some guidelines for the development of IHC diagnostic LDTs, it is unlikely that these will be stringent enough for the adequate validation of a PD-L1 biomarker assay that will be required to perform adequately over the dynamic range of staining and cover several different cut-offs [83, 84]. External quality assurance programmes will help laboratories achieve and maintain adequate performance.
Although there is considerable variation around the world, in the proportion of patients with advanced NSCLC for whom only cytology-type samples are available for diagnosis, it is not unusual for this proportion to be 50% or more. Currently, cytology samples are not trial validated for PD-L1 testing, but work is ongoing, trying to prove that these samples are adequate for testing [85]. There is some anecdotal evidence that PD-L1 staining is diminished by alcohol-based fixation, a common step in the preparation of many cytology-type samples.
Training is important in achieving good performance in reading PD-L1 IHC stains. The estimation of tumour proportion score can be quite challenging in some samples, particularly when immune cells, and especially macrophages, also express PD-L1. The sample used for scoring should have a minimum of 100 tumour cells, and all of the tumour present in the sample section should be used in the assessment. Studies of interobserver variability that have so far been reported describe moderate agreement, but this is often on a background of limited training and experience [77].
How should PD-L1 staining outcomes be reported? Although there might be a temptation to simply report cases as above or below a particular cut-off, as ‘positive’ or ‘negative’, it will surely be much better to give an actual figure for estimated tumour proportion score, as a percentage. Realistically this will be to the nearest 10% for cases showing more staining, but a more specific figure should be given if staining is under 10%. The report should indicate the details of the assay used and give some indication that the tumour cell number was adequate. If the SP142 assay is used, it is recommended to first assess the tumour proportion score. If this is below 1%, an immune cell score, expressed as the percentage area of tumour infiltrated by PD-L1 positive immune cells, is given. It is not yet clear what value there would be in attempting this assessment, or any other measure of immune cell staining, when reading any other assay.
Other Possible Biomarker Strategies for Immunotherapy in Lung Cancer
A number of other biomarker approaches are being investigated, in the search for a strategy that might either be better than PD-L1 IHC or, perhaps more likely, be used in conjunction with PD-L1 IHC to improve the efficiency of patient selection.
The importance of mutation burden, leading to neoantigenicity and possible immunogenicity, was discussed earlier in this chapter. A relatively high mutational burden, measured by whole-exome, next-generation sequencing, has been associated with superior response to pembrolizumab [86]. McGranahan and colleagues showed that it was important that the high mutation burden was a clonal phenomenon and not restricted to a subclone of cells in the tumour [87]. EGFR mutation is associated with poorer response, whilst KRAS mutation favoured response to nivolumab in non-squamous NSCLC in second or greater line therapy [64]. This probably reflects the fact that EGFR mutation is found in tumours not related to tobacco carcinogenesis and which have a lower overall mutation burden, whilst KRAS mutations are associated with smoking and tumours with a higher overall mutation burden. Similarly, smokers respond better than non-smokers to immunotherapy. Using whole-exome sequencing to deliver a routine, high demand biomarker would be challenging. One possible approach may be to find a smaller panel of mutations, perhaps including KRAS and TP53, which might be used as a surrogate for overall tumour mutation burden. There are interesting data beginning to emerge, relating different immune profiles in tumours, in association with different coexisting mutations in KRAS-mutated lung adenocarcinoma [88, 89]. High microsatellite instability (MSI high) similarly predicts for response in colorectal, endometrial and gastric carcinoma [90, 91], whilst polymerase E (POLE) mutations, also associated with DNA repair deficiency and high levels of genomic disarray, have been investigated in this context in other tumours [91]. These abnormalities are rare in lung cancer and unlikely to be useful.
For immunomodulatory therapy to work, a potential inflammatory response must exist. The immune system must be primed to recognise the immunogenic tumour cells. The therapy works by ‘releasing the brakes’ on the existing immune infiltrate, allowing specific cytotoxic T-cell action. As discussed above, an immune infiltrate in NSCLC may be prognostic, but could it be predictive of response to these drugs? None of the published trials of immunotherapy in lung cancer using drugs against the PD-1 axis present any data on the value of immune cell infiltrates, either in general or assessing particular cell types, as a predictive biomarker. In colorectal carcinoma and melanoma, however, there is some evidence that these infiltrates may help predict response [92]; as mentioned above, it is likely to be the ‘inflamed’ tumour with the adaptive immune response which is most likely to respond to immunomodulatory therapy [15, 92, 93].
Upregulation of genes related to the immune response and in particular gamma interferon function assessed in a tumour sample may well be an alternative way of identifying tumours with an immune infiltrate [94]. Immune gene expression signatures have been shown to select for a group of patients more responsive to atezolizumab in the Poplar trial [65]. A panel of around 14 serum cytokine levels was assessed in nivolumab -treated advanced NSCLC, where two, somewhat different, panels, when highly expressed in serum, were associated with better responses in squamous and adenocarcinoma, respectively [95]. High levels of these cytokines were also, however, associated with better outcomes in the chemotherapy arms of the trials, so there is the possibility that this biomarker is prognostic rather than predictive.
Other factors are related, or potentially related, to response to inhibitors of the PD-1 axis, but it is uncertain what role, if any, they may play in selecting patients for treatment [15]. Setting aside generic issues around performance status and other metabolic indicators like serum LDH, and a positive smoking history which is associated with responsiveness, the patient’s microbiome presents an intriguing matter worthy of exploration. The latter concerns the observation that immune responsiveness in general, and responses to immunomodulatory therapy in particular, may be influenced by the composition of the gut flora [96].
Currently, virtually all the biomarker data from clinical trials, published on NSCLC, concerns PD-L1 IHC. Trials are being designed with pre-specified selection of patients based upon PD-L1 expression, although many will have secondary analysis of other biomarkers. The introduction of pembrolizumab into first-line therapy for advanced NSCLC, with a PD-L1 IHC companion diagnostic, will consolidate the practice of PD-L1 IHC assessment in NSCLC [75, 76]. From the available evidence, it seems unlikely that an alternative biomarker approach will be found to replace PD-L1 IHC, in the context of selecting patients for PD-1 axis inhibitors; PD-L1 is, after all, either the target of these drugs or functionally inhibited by them. Most of the evidence suggests that, when PD-L1 is truly absent or at low levels in the tumour, these agents do not benefit the patient. It seems more likely that PD-L1 IHC will remain at the core of the testing process, but that additional biomarker approaches, such as some measure of mutational load and/or ‘inflammation’, may improve the overall selection process. How much additional improvement in ORR these strategies will achieve remains to be seen. How much improvement in ORR will be considered enough to justify an increasingly complex and costly testing process is also open to question. Some of the approaches mentioned above are certainly promising, but published tumour gene expression profiles are based upon a fresh, frozen tumour, a difficult medium for widespread, routine biomarker testing. Next-generation sequencing technology is still not a mainstream diagnostic tool, though things are improving, and this will undoubtedly become a reality in a majority of centres. Serum-based biomarkers are always attractive, given the ease of sample acquisition, but blood-based immune parameters are readily confounded by immune reactions unrelated to a cancer-specific response.
Conclusion
After many years of failed trials and major issues with toxicity, immunotherapy for advanced NSCLC has certainly come of age. Most of the failed approaches involved some attempt at stimulating the immune system, usually in a rather non-specific manner, and with toxic consequences for patients. Although a more specific, antigen-based, tailored approach involving the ex vivo production of tumour-specific T cells is being trialled, it seems that targeting immune-inhibitory mechanisms, to release an existing immune response, is a more successful strategy. Of course, this will not work for all patients with advanced NSCLC; for a minority their tumour will be non-immunogenic, and there is no effective immune response to reactivate. Increased understanding of the molecular mechanisms regulating the immune system, and how these processes interact with tumour development, not only in terms of cellular interactions but also at a molecular level, has been crucial in helping advance the cause of immunotherapy in lung cancer. An important driver of future developments, in both immunotherapy and the selection of patients for these treatments using biomarkers, will be our increasing understanding of the molecular pathology of lung cancer.
References
1.
Pennock GK, Chow LQ. The evolving role of immune checkpoint inhibitors in cancer treatment. Oncologist. 2015;20:812–22.CrossrefPubMedPubMedCentral
2.
Mountzios G, Linardou H, Kosmidis P. Immunotherapy in non-small cell lung cancer: the clinical impact of immune response and targeting. Ann Transl Med. 2016;4:268.CrossrefPubMedPubMedCentral
3.
Villaruz LC, Kalyan A, Zarour H, et al. Immunotherapy in lung cancer. Transl Lung Cancer Res. 2014;3:2–14.PubMedPubMedCentral
4.
Bagley SJ, Langer CJ. PD1/PD-L1 checkpoint blockade in non-small cell lung cancer. Clin Adv Hematol Oncol. 2015;13:676–83.PubMed
5.
Carbone DP, Gandara DR, Antonia SJ, et al. Non-small-cell lung cancer: role of the immune system and potential for immunotherapy. J Thorac Oncol. 2015;10:974–84.CrossrefPubMedPubMedCentral
6.
Ma W, Gilligan BM, Juan J, et al. Current status and perspectives in translational biomarker research for PD-1/PD-L1 immune checkpoint blockade therapy. J Hematol Oncol. 2016;27:47.Crossref
7.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64.CrossrefPubMedPubMedCentral
8.
Lynch TJ, Bondarenko I, Luft A, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: results from a randomized, double-blind, multicenter phase II study. J Clin Oncol. 2012;30:2046–54.CrossrefPubMed
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


