Immunology of Human Immunodeficiency Virus Infection
Douglas S. Kwon
Bruce D. Walker
INTRODUCTION
In June 1981, the U.S. Centers for Disease Control and Prevention (CDC) reported five cases of Pneumocystis pneumonia (PCP) in otherwise healthy young gay men in Los Angeles, California,1 representing the first published evidence of what was to become the acquired immunodeficiency syndrome (AIDS) epidemic. At that time PCP was rare, known as an “opportunistic infection” that occurred in the setting of severe immune compromise resulting from cancer treatment. At the same time, there were also reports of an aggressive form of Kaposi sarcoma, a generally benign cancer, in the same demographic, namely young gay men.2 Like PCP, Kaposi sarcoma was infrequent and was typically seen in older men or those who were immunosuppressed following organ transplantation. It made sense in the setting of severe immune compromise to see PCP or Kaposi sarcoma, but what was alarming in those initial reports was that it was being seen in otherwise seemingly healthy people.
At that time pentamidine, the main drug used to treat PCP, was only available through the CDC. Coincident with the first PCP cases reported in Los Angeles, there was a dramatic increase in requests for pentamidine from many other cities across the United States. A consistent and disturbing pattern was quickly noted—the drug was largely being requested for young men who had no known reason to be immune compromised and at risk for PCP. An emerging epidemic was thus recognized, and already from those first patients there were clues pointing to the central problem induced by what was ultimately recognized to be infection by a new human retrovirus: that human immunodeficiency virus (HIV) is an infection of the immune system.
When an antibody test for HIV became available in 1985, the kinetics of the expanding epidemic first began to be realized through retrospective examination of stored serum samples. Results from analysis of samples from a hepatitis B vaccine trial that had coincidentally been initiated in 1980 in gay and bisexual men in San Francisco, California, were startling.3 By 1982, the cumulative incidence of infection in those at-risk men had risen to 42.6%. HIV has since gone on to infect over 60 million people worldwide and has caused over 30 million deaths.4 In some of the most affected areas of the world, HIV prevalence exceeds 50% in certain communities and age groups (Fig. 42.1).5
Thankfully, the development of effective HIV drug therapy has turned HIV infection into a treatable condition for those fortunate to have access to these medications,6 but viral integration into the host chromosome makes the need for treatment lifelong.7 Regrettably, for each person who begins antiretroviral therapy (ART) in resource-limited settings, there are two who become newly infected.4 The ultimate resolution of the epidemic will almost certainly require an effective vaccine, but despite nearly a billion dollars invested annually in AIDS vaccine development, this has remained an elusive goal. Unlike other infections for which effective vaccines exist, for HIV there is a lack of natural clearance and subsequent durable protection.8 As natural protective immunity does not exist, an effective HIV vaccine will have to do better than nature does.
HUMAN IMMUNODEFICIENCY VIRUS ORIGIN, TRANSMISSION, AND DISEASE PROGRESSION
Origin of Human Immunodeficiency Virus and Relationship to Other Retroviruses
The isolation of a retrovirus in 1983 from a patient with AIDS in France led to wider sampling of at-risk persons and was paralleled by the discovery of other primate retroviruses. Sequencing of HIV revealed enormous variability among isolates, which were ultimately phylogenetically characterized as belonging to four distinct groups. Group M is the largest and accounts for over 98% of infections worldwide.9 Based on geographically targeted sequencing, it appears that the epicenter of the global group M epidemic was in Kinshasa in the Democratic Republic of the Congo,10 and that a cross-species transmission of simian immunodeficiency virus (SIV) from chimpanzees occurred in the early 1900s.11,12 In turn, chimpanzees had acquired SIV from cross-species transmission of two viruses in monkeys that recombined. The actual origin of the group M epidemic in humans appears to have been in a remote region of southeastern Cameroon, likely by exposure through butchering of a chimpanzee.13 Much less frequent are groups N, O, and P, the result of separate cross-species transmissions from chimpanzees (groups N and O) and gorillas (group P), which have remained localized to Cameroon and neighboring countries in west Africa.9,14,15
HIV is a typical retrovirus, morphologically characterized by a core containing the viral ribonucleic acid (RNA) and two copies of reverse transcriptase (RT), surrounded by an outer envelope that is heavily glycosylated. The errorprone RT leads to the rapid generation of new mutations, resulting in enormous genetic variation within group M viruses globally. This is largely due to lack of 3′ exonuclease proofreading activity of RT, which gives rise to the mutation of approximately 1 in every 10,000 nucleotides during viral genome transcription; in other words, once per replication
cycle16. Additional factors are the frequent viral recombination and the lifelong nature of infection. Within an individual, this produces an enormously diverse quasispecies of viruses whose intraindividual diversity exceeds the global diversity of influenza in a single season17 (Fig. 42.2). This represents a significant challenge to the immunologic control of infection and the development of broadly prophylactic vaccines. Currently, there are at least nine distinct clades, or subtypes, of group M HIV-1 worldwide, and many additional subtypes that are the result of recombination events between these major clades.
cycle16. Additional factors are the frequent viral recombination and the lifelong nature of infection. Within an individual, this produces an enormously diverse quasispecies of viruses whose intraindividual diversity exceeds the global diversity of influenza in a single season17 (Fig. 42.2). This represents a significant challenge to the immunologic control of infection and the development of broadly prophylactic vaccines. Currently, there are at least nine distinct clades, or subtypes, of group M HIV-1 worldwide, and many additional subtypes that are the result of recombination events between these major clades.
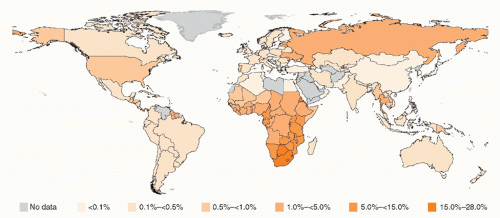 FIG. 42.1. Global Human Immunodeficiency Virus Prevalence by Country. From World Health Organization.4 |
HIV-1 is closely linked to HIV-2, a less pathogenic human retrovirus that is endemic in west Africa. HIV is also closely related to SIVs, a number of nonhuman primate retroviruses.18 Although SIV was initially considered nonpathogenic in primates, infection of certain wild chimpanzee species results in CD4 T-cell decline and increased mortality. It is important to note that some wild nonhuman primate species tolerate their species-specific SIV infections quite well, including African green monkeys and sooty mangabeys, in whom the infection is characterized by persistent high viral load but a lack of CD4 cell decline or development of immune deficiency.19
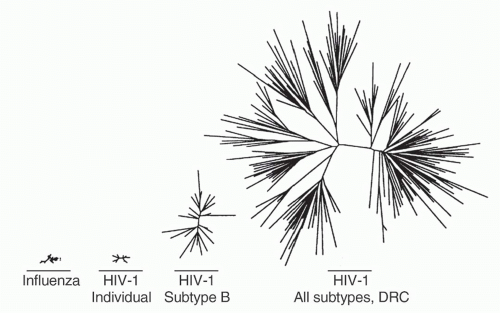 FIG. 42.2. Relative Viral Diversity of Influenza and Human Immunodeficiency Virus (HIV)-1. Comparison of influenza HA1 domain and HIV-1 envelope V2-C5 regions, starting from left with diversity of all H3N2 influenza sequences available from the 1996 influenza season, then HIV-1 subtype B sequence diversity from a single individual, HIV-1 sequences from a subtype B epidemic occurring in Amsterdam in 1990 to 1991, and finally HIV-1 sequences from the Democratic Republic of Congo obtained in 1997. Adapted from Korber et al.17 |
Exploiting Knowledge of the Viral Life Cycle to Develop Antiviral Agents
Early in the HIV epidemic, it was realized that expression of CD4 was necessary for cells to become infected—a finding that was assisted by the recognition that CD4+ cells were specifically lost in progressive disease. However, it was also clear that CD4 alone was not sufficient. Expression of recombinant CD4 on murine cells failed to allow viral infection. The discovery of chemokine receptors as necessary coreceptors for HIV entry took over a decade to be realized and was facilitated by a remarkable convergence of clinical observation and basic science. Blood samples from persons identified to be very heavily exposed to HIV, who had never become infected, showed that a group of β chemokines: MIP-1α, MIP-1β, and RANTES, were secreted from CD8 T cells and could inhibit HIV replication.20 These chemokines were known to be linked through a common receptor, CCR5, a member of a family of G-protein coupled seven transmembrane domain chemokine receptors. In the next year, CCR5 and another chemokine receptor, CXCR4, were identified as the long-sought HIV entry coreceptors whose expression along with CD4 were both necessary and sufficient to allow viral entry.21,22,23 Shortly thereafter, it was discovered that homozygosity of a naturally occurring 32 base pair deletion of
CCR5 (CCR5 δ32) that abrogated surface expression of the protein and occurred in 1% of Caucasians imparted resistance to HIV acquisition, thus demonstrating the biologic significance of chemokine receptors for HIV infection.24,25,26 As individuals lacking functional CCR5 were found to be largely immunologically intact, CCR5 became an attractive target for ART; in 2007, maraviroc, a negative allosteric modulator of the CCR5 receptor, became the first chemokine receptor antagonist approved for treatment of HIV infection.
CCR5 (CCR5 δ32) that abrogated surface expression of the protein and occurred in 1% of Caucasians imparted resistance to HIV acquisition, thus demonstrating the biologic significance of chemokine receptors for HIV infection.24,25,26 As individuals lacking functional CCR5 were found to be largely immunologically intact, CCR5 became an attractive target for ART; in 2007, maraviroc, a negative allosteric modulator of the CCR5 receptor, became the first chemokine receptor antagonist approved for treatment of HIV infection.
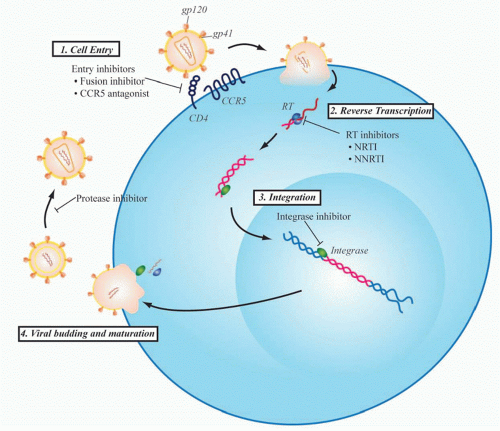 FIG. 42.3. Human Immunodeficiency Virus (HIV) Viral Life Cycle and Targets for Antiretroviral Therapy. The HIV envelope protein gp120 first contacts CD4 on the target cell, which then induces conformational changes in gp120 that exposes the chemokine receptor binding site. Following binding of chemokine receptor, HIV gp41 mediates fusion of target and viral membranes. The viral single-stranded ribonucleic acid genome is then reverse transcribed, and the viral genome is transported to the nucleus where it is integrated in the host genome. Viral genes are then transcribed and translated, and virions then bud from the host membrane. Following budding, virions mature in a process that requires cleavage of HIV polyproteins by the viral protease. Currently approved antiviral agents target entry, reverse transcription, viral genome integration, and viral protease function. |
Even before the development of maraviroc, however, multiple viral proteins had been successfully targeted to treat HIV infection based upon a rapidly developing understanding of the viral life cycle (Fig. 42.3). HIV RT was the first viral protein to be successfully targeted by drug therapy, and the class of RT inhibitors remains the backbone of firstline regimens of combination ART strategies used today. Zidovudine was the first clinically approved HIV drug. It is a thymidine analog initially developed in 1964 as a potential anticancer agent. Over 20 years later, however, it was found to inhibit replication of HIV as part of a unique collaboration between the National Cancer Institute and the pharmaceutical company Burroughs Wellcome. When zidovudine was first tested for activity against HIV in 1985, there were over 10,000 people suffering from AIDS, a disease for which there was no available therapy at that time.27 After proving to be safe in a small phase I trial, zidovudine quickly went on to efficacy studies and remarkably went on to be approved for clinical use by the U.S. Food and Drug Administration just 2 years after its first testing in the laboratory. This would be the first of several drugs that target RT that now constitute one of the mainstays of HIV treatment.
The next major breakthrough in ART was the development of drugs that target the HIV protease. Protease is required for cleavage of viral polyproteins in immature virions released from the infected cell, a step necessary for virion assembly and maturation. The first protease inhibitor, saquinavir, was approved in 1995, and its development marked the age of modern multidrug ART. Up to that point, only four drugs had been developed, all nucleoside RT inhibitors (NRTIs). However, therapy with these NRTIs showed toxicity, and none of them alone or in combination could durably control HIV. Combination therapy with agents that inhibited both RT and protease showed strong in vitro synergy,28 could achieve virologic suppression in vivo, and resulted in a dramatic decrease in AIDS deaths (Fig. 42.4).29 This was quickly followed by the development of non-NRTIs, which also target RT and can also be used effectively with NRTIs to suppress viral replication. This marked the
modern era of combination ART and has changed HIV from a nearly uniformly terminal illness to a chronic treatable disease for those with access to medications.
modern era of combination ART and has changed HIV from a nearly uniformly terminal illness to a chronic treatable disease for those with access to medications.
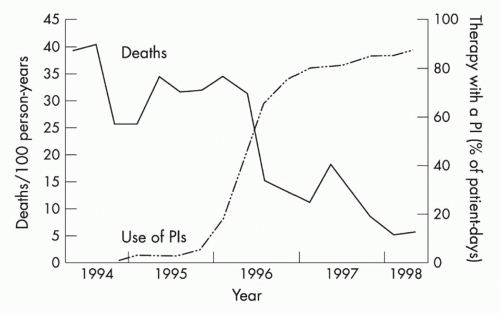 FIG. 42.4. Protease Inhibitors and Decline in Acquired Immunodeficiency Syndrome (AIDS) Mortality. Mortality from AIDS showed a significant decrease following the introduction of protease inhibitors, which were used in combination with agents that target human immunodeficiency virus (HIV) reverse transcriptase to treat HIV-infected individuals with combination therapy to suppress viral replication. From Palella et al.29 |
Since the development of RT and protease inhibitors, two other steps in the viral life cycle, entry and viral genome integration, have been successfully targeted by approved drugs. HIV entry is initiated when the viral envelope protein, gp120, contacts CD4 and a chemokine receptor on the target cell membrane. This triggers conformational changes in the transmembrane envelope glycoprotein, gp41, which leads to fusion of viral and host membranes. There are currently two antiretroviral agents that act at this early stage of entry: maraviroc, a CCR5 antagonist that blocks the interaction of gp120 with CCR5, and enfuvirtide (Fuzeon, T-20), approved in 2003, a 36 amino acid peptide that binds to gp41 and prevents formation of a hairpin intermediate necessary for fusion of the viral and target cell membranes. Raltegravir targets HIV integrase and represents the newest class of antiretroviral agents to be developed. Integrase is the viral enzyme that catalyzes the deoxyribonucleic acid strand transfer reactions necessary for incorporation of the viral genome into the host genome. It represents one of the most potent of currently available HIV drugs and is generally one of the best tolerated therapies.
Although there are currently over 30 approved antiretroviral drugs in various formulations, demand continues for novel agents, especially as drug resistance becomes increasingly common. Studies in Europe have demonstrated that approximately 10% of all newly diagnosed patients harbor virus with at least one major resistance-associated mutation.30 New drugs remain in development including maturation inhibitors, CD4 receptor inhibitors, and additional agents in existing classes.31 The development of suppressive multidrug ART has saved millions of lives and has only been possibly with a detailed understanding of the molecular mechanisms of HIV pathogenesis. These drugs are now being used not only to treat those who are infected, but also are being tested for use in preventing infection in individuals at high risk of acquisition.32
Mucosal Transmission and Early Local Viral Replication
Transmission of HIV is relatively inefficient with a risk of approximately 1 in 1,000 sexual acts for male-to-female sexual transmission and 1 in 100 for receptive anal intercourse.33 Over 90% of HIV transmissions worldwide occur across a mucosal barrier, either through sexual exposure or maternalto-child transmission, with the remaining cases due to direct blood exposure such as injection drug use. These mucosal surfaces are exposed to an enormously diverse quasispecies of genetically distinct virions, and yet the majority of transmissions are mediated by a single founder virus,34 although as many as 16 viruses have been documented to be transmitted simultaneously.35 Multiple transmitting founder viruses appear to be more prevalent in transmission occurring via intravenous drug use, suggesting that mucosal-specific factors are an important determinant for this restriction.35 Following mucosal exposure, a small founder population establishes host infection by initially replicating locally within the exposed mucosal tissue for 7 to 10 days before disseminating to the draining lymph node and then moving quickly into the blood.36 Once infection has spread to the periphery, a widespread reservoir of latently infected cells is rapidly established at multiple sites and subsequent viral eradication becomes enormously challenging due to this extensive reservoir.
The fact that HIV transmission is inherently inefficient and typically initiated by a single virion that requires days of local expansion before systemic infection can take place suggests that these early events represent a vulnerable stage in the process of transmission. Unfortunately, few HIVspecific B- or T-cell responses are detected locally in the mucosal site of exposure until well after viral dissemination, despite the fact that these sites contain abundant antigenpresenting cells and numerous lymphocytes.37,38 This lack of early responses likely plays a critical role in the failure of the immune system to contain viral replication within the genital mucosa. Although some studies suggest that a subset of persons with multiple exposures will develop adaptive immune responses to HIV,39 and have suggested that these can protect against acquisition, this remains controversial.40
Transmission depends on specific properties of the envelope glycoprotein of HIV. Acute infection is typically established with R5 viruses that utilize CCR5 as a coreceptor. Over time, some viruses within the quasispecies of an infected person can evolve to an X4 phenotype, which is characterized by the use of CXCR4 as its coreceptor, but these viruses somehow are either prevented from establishing the initial infection or rapidly revert to R5 viruses upon transmission.
The mechanism of the transmission “bottleneck” remains incompletely understood. The efficiency of HIV transmission is actually quite low and highly linked to the viral load in the donor. A population study of 45 discordant couples, in which one partner is HIV positive and the other is not, showed a complete lack of transmission in the 15 couples in whom the infected partner had a viral load of < 1,500 RNA copies/uL plasma.41 More recently, treatment of couples discordant for HIV infection has confirmed the impact of viral load on transmission: when the HIV-infected partner is on therapy, the risk of HIV transmission to his or her partner is reduced by 96%.42 Periods of highest transmissibility tend to be the acute phase of infection, before viral load has declined to a steady state, and end stage disease, when viral load again rises.43 Importantly, the highest viral loads are seen in acute infection, before antibodies become detectable, such that standard enzyme-linked immunosorbent assay antibody testing is typically negative at the time of highest risk of transmission. This is being addressed at least in part through next generation tests that detect both antibody as well as p24 antigen, decreasing the “window period” when persons are infected but test negative.
Acute Infection Syndrome and Early Infection
Most persons have had the symptoms that could be consistent with acute HIV infection at some point in their lives as these are not specific to HIV but rather are typical of many viral infections. Indeed, the diagnosis of acute HIV infection is frequently missed by health care providers because at the time of acute symptoms, anti-HIV antibodies have not yet
developed and thus persons test negative by the standard antibody tests that are used to diagnose infection. Most but not all patients are symptomatic with their initial acute infection, with fever, sore throat, swollen lymph nodes, fatigue, malaise, and often transient oral or genital ulcers.44 The syndrome looks very much like acute infectious mononucleosis.
developed and thus persons test negative by the standard antibody tests that are used to diagnose infection. Most but not all patients are symptomatic with their initial acute infection, with fever, sore throat, swollen lymph nodes, fatigue, malaise, and often transient oral or genital ulcers.44 The syndrome looks very much like acute infectious mononucleosis.
The acute symptoms, which are nonspecific, eventually resolve, and subsequent disease course is characterized by a gradual CD4 cell decline and slowly rising viral load, with the ultimate development of clinical AIDS when the CD4 count is < 200 cells/uL or certain AIDS-defining illnesses arise.
Initial peak viremia in newly infected adults is often 10 million viral particles per mL or more,45 followed by a decline over the first few months to a quasisteady state at a mean of around 30,000 RNA copies/mL (Fig. 42.5).46
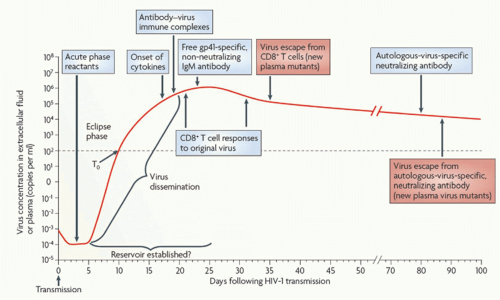 FIG. 42.5. Early Immune Responses in Acute Human Immuno deficiency Virus (HIV)-1 Infection. Acute infection is typically characterized by high viral loads and flu-like symptoms. There is an initial “eclipse phase” in which virus is present but antibody-based HIV tests remain negative. Early adaptive responses are comprised of HIVspecific CD8 T-cell responses and nonneutralizing antibody responses. The emergence of CD8 T-cell responses coincides with the decline of peak viral loads and the establishment of a viral set point. From McMichael et al.190 |
Current views of the very earliest stages in acute infection stem largely from studies in the nonhuman primate model of AIDS virus infection, in which rhesus macaques are infected with SIV. In high-dose challenge models of macaques, virus crosses the mucosal site of exposure within hours and initially establishes a small founder population of infected cells locally.47 It is unclear how the virus crosses the mucosal epithelial barrier, but studies have suggested a variety of mechanisms, including transcytosis across epithelial cells, facilitation by mucosal dendritic cells (DCs), and/or entry through mechanical disruption or physiological intercellular spaces in the epithelium. Once across, the first cells to be infected are CD4 T cells. Surprisingly, though HIV preferentially infects activated CD4 cells, the first targets appear to be resting CD4 T cells, or cells that have been previously activated and still express CCR5 coreceptor but have entered into a more quiescent state. Once infection within the tissue is established, there is a period of local expansion that lasts for 7 to 10 days before the virus is disseminated to regional lymph nodes. This is then followed by rapid progression to the peripheral circulation and seeding of gut-associated lymphoid tissue (GALT). This gut reservoir contains 60% to 80% of the body’s CD4 T cells, a large majority relative to the 2% to 3% of lymphocytes present in the peripheral circulation.
Based upon studies initially conducted in macaques and subsequently corroborated in humans, a hallmark of HIV infection is the rapid and profound depletion of GALT CD4 T cells in the lamina propria within the first weeks of HIV infection (Fig. 42.6). It appears that these cells never fully recover, even with prolonged treatment with antiviral medications that suppress peripheral viral loads and result in
rebound of CD4 T-cell counts in the peripheral blood.48,49 This CD4 T-cell depletion is accompanied by extensive apoptosis of bystander cells, associated immune activation, and disruption of normal epithelial barrier function in the gut. Recent data suggest that bystander death actually involves abortive infection, with accumulation of cytoplasmic nucleic acids that elicit proapoptotic and proinflammatory responses.50
rebound of CD4 T-cell counts in the peripheral blood.48,49 This CD4 T-cell depletion is accompanied by extensive apoptosis of bystander cells, associated immune activation, and disruption of normal epithelial barrier function in the gut. Recent data suggest that bystander death actually involves abortive infection, with accumulation of cytoplasmic nucleic acids that elicit proapoptotic and proinflammatory responses.50
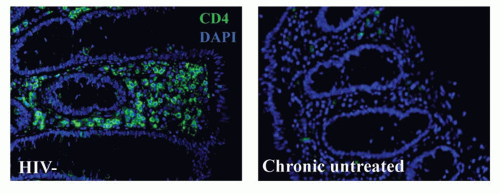 FIG. 42.6. Depletion of CD4 T Cells in the Intestinal Mucosa. Gut-associated lymphoid tissue contains the majority of the body’s lymphocytes, which reside within both the lamina propria and lymphoid follicles. Lamina propria CD4 T cells are rapidly and massively depleted early in human immunodeficiency virus (HIV) infection. Shown are representative stains from colonic mucosa from HIV-infected and -uninfected individuals stained by immunofluorescence for CD4 (green) and nuclei (4′,6-diamidino-2-phenylindole [DAPI], blue). |
Natural History of Untreated Infection
The typical course of untreated HIV infection until the development of AIDS, defined as a CD4 count of < 200 cells/µL or the presence of certain AIDS defining illnesses is approximately 10 years51 and involves progressive immunologic decline. As the epidemic has matured, however, it has become clear that not all persons progress. There are now persons with documented untreated infection for more than 30 years who have maintained normal CD4 cell numbers and an undetectable viral load, whereas others progress from acute infection to AIDS in as little as 6 months without therapy.52
The best HIV natural history data derive from the Multicenter AIDS Cohort Study and other population studies established in the early days of the epidemic. The Multicenter AIDS Cohort Study, which involved recruitment of both infected and at risk men in Baltimore, Maryland, Chicago, Illinois, Los Angeles, California, and Pittsburgh, Pennsylvania, was established in 1984 and continues today. The study currently has almost 7,000 participants with an accumulated 87,000 person years of follow-up,53 with a large repository of archived specimens. These multicenter seroprevalent and seroincident cohorts were pivotal in identifying laboratory predictors of prognosis, including CD4 T-cell count and viral load, which are both independent predictors of disease progression. Following acute infection, viral load reaches a quasi-set point within approximately 6 months of infection, with a median viral load around 30,000 RNA copies/mL and interquartile ranges from 12,000 to 80,000 copies/mL. There is a marked increase in the development of opportunistic infections when CD4 count drops below 200 cells/µL, which led to the current recommendations that prophylactic treatment of opportunistic infections begin when this level is reached. The sequelae of chronic untreated HIV infection are the development of these opportunistic diseases, which are what led to the original identification of the new epidemic of HIV infection. What was being seen clinically was the end stage of a chronic infection in which immune dysfunction allowed the establishment of a number of otherwise rare illnesses.
Among the CDC AIDS-defining illnesses are candidiasis of the respiratory tract, Pneumocystis jiroveci pneumonia, cytomegalovirus disease, extrapulmonary cryptococcal disease, toxoplasmosis of the brain, and mycobacterial infection,54 all indicative of an underlying defect in cellular immunity. Mycobacterium tuberculosis is a particularly devastating coinfection, in part because its widespread prevalence, with almost one-third of HIV-infected individuals globally also infected with tuberculosis. Both infections are far more prevalent in resource-limited settings, and coinfection is associated with worse outcomes, with tuberculosis being the leading cause of death in coinfected individuals.55 Although opportunistic infections are a hallmark of progressive HIV, AIDS-defining illnesses are not confined to infectious diseases. There are notably several cancers that are also associated with HIV, including Kaposi sarcoma, lymphoma, and cervical cancer, all of which are likely due to loss of protective immune surveillance and the presence of other viral coinfections. Additionally, although not listed by the CDC as AIDS defining, there is also evidence that HIV-infected individuals are at increased risk for anal cancer, hepatocellular carcinoma, and lung cancer.56 The risks for associated infectious diseases decrease with therapy and subsequent immune reconstitution; however, the risk for some cancers, such as cervical and anal cancer, remains elevated.
With the advent of combination ART, HIV has in many ways become a chronic treatable disease. However, despite the immunologic and virologic benefits of ART, life expectancy in HIV-infected individuals remains approximately 14% shorter than the average for uninfected persons.57 The ART Cohort Collaboration study, a group composed of 13 independent studies, found that deaths were due largely due to age-related clinical diseases, such as cardiovascular disease, cancer, fractures, and renal dysfunction, despite apparent immune reconstitution with effective ART. Additionally, treated HIV-infected adults older than 50 years old had three times greater risk of death compared to those younger than 30 years. The precise mechanism of this age-related morbidity and mortality is unclear, but it is increasingly relevant as the population of HIV-infected individuals ages, a result of the success of ART. It is projected that by 2015 more than half of all HIV-infected individuals in the United States will be over 50 years of age,58 and given their increased risk of morbidity and mortality despite what is considered successful treatment, the mechanism of these age-related diseases is an area of critical research. One key to understanding this is the observation that HIV-infected individuals have increased systemic immune activation and that with therapy the activation level decreases but does not reach the same state of those who are uninfected. Persistent immune activation, even at low levels, may therefore be one mechanism by which HIV creates progressive impairment.
Chronic Immune Activation and Immune Dysregulation
Peripheral markers of T-cell activation predict the rate of disease progression and CD4 T-cell decline in untreated infection more accurately than plasma viral load.59,60,61 This persistent immune activation is a hallmark of chronic HIV infection; although it is clear that HIV viremia itself plays a direct role, it does not fully account for the entirety of observed T-cell activation seen in infected patients. For example, in sooty mangabeys, a natural host of SIV, high levels of viremia are supported but without a significant increase in immune activation and with the remarkable absence of clinically advanced disease.62,63,64 Additionally,
patients who are successfully treated with ART and have suppressed viral loads continue to have elevated markers of T-cell activation relative to HIV-uninfected subjects.65
patients who are successfully treated with ART and have suppressed viral loads continue to have elevated markers of T-cell activation relative to HIV-uninfected subjects.65
Recent studies have suggested that the gut may be critically involved in chronic immune activation. Individuals with untreated HIV infection have been found to exhibit elevated serum levels of bacterial lipopolysaccharide (LPS), which is thought to be a consequence of increased microbial translocation from the intestinal lumen in the setting of a damaged gut mucosa.66 Serum LPS levels are closely correlated with markers of systemic T-cell activation; in patients who receive allogeneic bone marrow transplants complicated by graft-versus-host disease, it has been shown that plasma LPS levels closely correlate with biochemical and histologic measures of mucosal damage.67 Thus, elevated serum LPS in HIV-infected subjects is believed to reflect the loss of mucosal epithelial integrity concomitant with HIV infection, and it defines a mechanism by which the gut may play a central role in chronic immune activation through increased microbial translocation. The ultimate anatomic cause of this increased translocation remains to be fully defined The ultimate consequence of this chronic immune activation appears to be higher turnover of CD4 and CD8 T cells, clonal exhaustion, and the progressive impairment of T-cell function.
The development of immune dysfunction or “exhaustion” in the presence of ongoing antigen exposure is a cardinal feature of chronic viral infections with persistent viremia.68,69 Exhaustion results in the hierarchical impairment of T-cell effector functions, with an initial loss of proliferative capacity and interleukin (IL)-2 production and the late loss of the ability to secrete proinflammatory cytokines such as interferon (IFN)γ.70 This exhaustion is mediated by several immunoregulatory pathways,69,71 including programmed death 1 (PD-1), cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), T-cell immunoglobulin and mucin-3 (TIM-3), and IL-10, which are all upregulated in vivo in chronic untreated HIV infection and have been shown to mediate a reversible virus-specific immune dysfunction in vitro. These immunoregulatory pathways are therefore being explored as potential targets for therapeutic intervention in chronic HIV infection.69,72,73,74
IMMUNE RESPONSE TO HUMAN IMMUNODEFICIENCY VIRUS
Unlike many other viral infections, there does not appear to be natural clearance of HIV infection. The only known case in which virus has been apparently successfully eliminated occurred in a patient who underwent a bone marrow transplant from a donor who was homozygous for the 32 base pair deletion in CCR5, rendering the newly transplanted cells not susceptible to infection by the R5 virus strain with which he was infected.75 The fact that people do not spontaneously clear virus indicates by definition that natural immunity is insufficient to eliminate HIV. Despite this, there is clear evidence of initial partial control in the early stages of infection, when viral load is decreased from peak levels often in excess of 10 million viral particles per mL of plasma to a set point mean of around 30,000 copies/mL.45 Moreover, some rare individuals termed elite controllers maintain undetectable viral loads and normal CD4 cell counts, in some cases for over 30 or more years, suggesting that partial host control is possible. Evidence exists to implicate both innate and adaptive immune responses in situations of partial control in acute and chronic infection, but the actual correlates of protective immunity remain unclear.
Natural Killer Cell Response
The potential protective role of natural killer (NK) cells in human viral infections is clear from examples of genetic defects in NK cells. For example, severe herpes simplex virus infection was observed in an adolescent with no NK cells.76 Aside from the fact that NK cells would be the first possible line of defense against HIV, HIV downregulates human leukocyte antigen (HLA)-A and -B molecules on infected cells, which would render them targetable by certain NK-cell subsets. NK-cell interactions with HIV-infected cells do not depend on expression of an antigen-specific receptor, but instead are dependent on alterations in activating and inhibitory signaling between HIV-infected cells and NK cells. This occurs via interactions with a subset of highly polymorphic killer immunoglobulin-like receptors (KIRs) with HLA class I molecules, as well as through indirect mechanisms involving antibody-dependent cellular cytotoxicity and an arsenal of nonspecific inhibitory and activating receptors.77,78,79
The evidence for a beneficial effect of NK cells in HIV infection is mounting, both in protection from acquisition and in subsequent control of viremia. A potential functional role for NK cells in blocking viral acquisition is supported by studies in exposed, uninfected persons who appear to be protected from infection. Persons who have been heavily exposed to HIV and yet remain uninfected have increased NK cell activity80 and a higher KIR3DS1 to KIR3DL1 transcript ratio,81 consistent with the genetic studies indicating a protective effect of the compound genotype consisting of the activating KIR KIR3DS1 and Bw4-80I.78 Studies in persons who are acutely infected also support an antiviral role for NK cells. There is an expansion of KIR3DS1+ NK cells in acute infection in persons expressing the putative ligand Bw4-80I, and these cells are able to lyse HIV infected cells in vitro, in a Bw4-80I-dependent manner.82
The mechanism of putative protection by NK cells in HIV infection likely relates to the selective downregulation of HLA A and B alleles by HIV. This certainly would be consistent with the role of activating ligands, but the inhibitory KIR allelic association is more challenging to explain. It may relate to the strength of the KIR-HLA interaction during NK-cell licensing, when stronger engagement of inhibitory KIRs by HLA molecules enhances their potential functional capacity. Stronger inhibitory signals during NK-cell development could lead to greater activation when infected cells downregulate HLA-A and -B, thus leading to enhanced function.78
There are a number of defects in NK cells that have been identified in HIV-infected persons. There is an inverse correlation between viral load and NK cell-mediated suppression of viral replication, suggesting that NK-cell dysfunction
may contribute to disease progression.83 NK cells in HIVinfected persons have impaired cytotoxic capacity associated with decreased levels of granzyme A and perforin,84 whereas other studies suggest that NK cells may be incompletely activated in chronic HIV infection and that functionally anergic subsets may be expanded.85 It has also been suggested that NK cells can become HIV-infected and might contribute to the persistent HIV reservoir in vivo.86
may contribute to disease progression.83 NK cells in HIVinfected persons have impaired cytotoxic capacity associated with decreased levels of granzyme A and perforin,84 whereas other studies suggest that NK cells may be incompletely activated in chronic HIV infection and that functionally anergic subsets may be expanded.85 It has also been suggested that NK cells can become HIV-infected and might contribute to the persistent HIV reservoir in vivo.86
More recently, in vivo evidence for antiviral selection pressure mediated by NK cells has been reported. Functional roles for NK cells are suggested by studies in infected persons, showing expansion of a subset of NK cells in acute infection.82 But the most compelling data implicating NK cells in the host defense against HIV come from the identification of NK-cell “footprints” in viral sequences obtained from infected persons. Analysis of KIR genotypes and fulllength virus sequencing in a large cohort of persons with chronic HIV infection demonstrated 22 positions throughout the HIV genome in which amino acid polymorphisms were associated with the presence of specific KIR genes.87 In vitro studies showed that these HIV sequence polymorphisms increased the binding of inhibitory KIRs to HIVinfected cells, suggesting a functional consequence favoring the virus. Moreover, these polymorphisms reduced the antiviral activity of KIR-positive NK cells, indicating not only that NK cells exert immune selection pressure on HIV, but also that HIV evolves to escape from detection by this response.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


