by those that studied innate immunity, that the adjuvant effects of microbial products like LPS were not mediated by specific receptors. Janeway articulated the need for a collective mea culpa among adaptive immunologists in 1989, describing the attitude to microbial adjuvants as “the immunologist’s dirty little secret,”9 and went on to hypothesize a parallel set of pathogen-associated molecular pattern receptors.10 The question of how LPS, and by extension many microbial adjuvants, were recognized by the vertebrate immune system was answered dramatically with the discovery of precise mutations in the signaling domain of a receptor that looked nothing like antibody, toll-like receptor (TLR) 4, in strains of mice that were specifically unable to respond to LPS.11 Mouse molecular genetics subsequently illuminated specific sensing functions for a diverse family of vertebrate TLRs and other receptors for “microbe markers.”12
T cells, which are indeed biased to be weakly self-major histocompatibility complex (MHC) reactive. Many of these polyreactive receptors do not pose a risk of autoimmune disease unless made in extraordinary concentrations, and when expressed in transgenic mice some have an affinity for self that falls below that which invokes actively acquired tolerance, as described later. But the assays used at the time could not distinguish between lymphocytes bearing frequent but harmless polyreactive receptors and much rarer cells bearing potentially destructive receptors with higher affinity and specificity, which we know now are usually controlled by active tolerance mechanisms. In other cases, highaffinity receptors were isolated but the assays used confused genuine autoantibodies with antibodies that bound to misfolded, proteolytically cleaved, or posttranslationally modi-fied “altered self.”29
I-J molecule,45,46 and antigen-specific suppressor T-cell clones were found to lack a T-cell receptor (TCR).47 While molecular genetics swept I-J+ CD8+ suppressor T cells away in the 1980s, in 2001 these same powerful experimental methods gave back CD4+ Foxp3+ “regulatory T (Treg) cells” and showed these were critical for tolerance, when Ramsdell and colleagues revealed that deficiency of the Foxp3 transcription factor unleashed lethal autoimmunity in the scurfy mutant mouse and in human immune dysregulation polyendocrinopathy enteropathy X-linked (IPEX) syndrome.48,49,50
that superantigens bind the sides of MHC II and TCR V&bgr; segments in an unconventional way that does not involve the conventional peptide-binding groove of MHC and complementarity determining regions of the TCR.
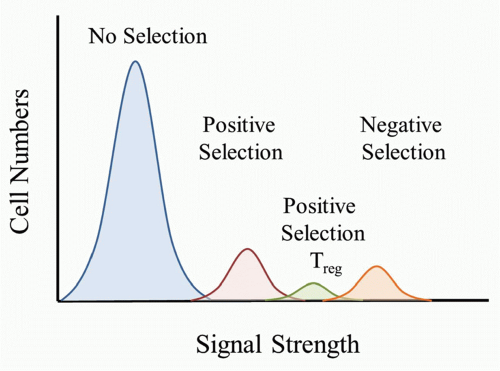 FIG 32.1. The Strength of T-Cell Receptor (TCR) Peptide/Major Histocompatibility Complex (MHC) Interactions Programs Tolerance Mechanisms. The random rearrangements of TCR gene segments lead to a wide range of specificities. Thymocyte fate will be determined by the strength of interaction between the TCR and peptide/MHC complexes that are found in the thymus. The majority of cells will not meet the threshold for positive selection. Stronger TCR interactions lead to the maturation of regulatory T cells or T-cell tolerance resulting in anergy or clonal deletion. The relative proportion of selected T cells is estimated. |
self-peptide/MHC complexes (pMHC) would be destined to die because it would be a “useless” TCR to have in the repertoire. TCRs that bound only weakly to self-pMHC complexes in the thymus would send a signal sufficient for the T cell to survive and differentiate to either the CD4 or CD8 lineage. According to this model, a strong signal would be sent if the TCR bound strongly to self-pMHC complexes and would lead to the death of the T cell. It is important to keep in mind that as the thymocyte undergoes positive selection and matures, the TCR level increases, and therefore the T cell would have increased sensitivity to self-pMHC complexes as they progress through thymocyte selection.
inbred rodent strains led to the development of autoimmunity targeting the gonads and thyroid gland.112,113,114 Further studies using various markers identified a subset of cells, first in the rat and then in the mouse, that had the potential to block the induction of autoimmunity. Mason’s team demonstrated that adoptive transfer of naïve T cells to immunodeficient rats resulted in autoimmune diabetes and inflammatory bowel disease that was blocked if a subset of CD4 T cells with markers of activated or memory T cells was cotransferred.115,116 These findings were extended to mice by Powrie117 and by Sakaguchi, whose group demonstrated that the transfer of CD4+CD25- cells into 6-week-old BALB/c nude mice resulted in the development of autoimmune oophoritis, gastritis, and thyroiditis that was suppressed by the cotransfer of CD4+ CD25+ cells.118 While these experiments suggested the existence of a new type of suppressor T cell (dubbed a Treg cell to avoid confusion with the CD8+ suppressor T cells of the 1980s), their interpretation was uncertain: suppression by cells with markers of activated or memory T cells could simply reflect better control of microbial flora in the immunodeficient recipients, so that there was less stimulus for inflammatory bowel disease and autoimmunity.
negative selection, thereby supporting the importance of this molecule.160 Therefore, studies support a role for ERK5 and Nur77 in thymocyte clonal deletion.
(APECED) or autoimmune polyendocrinopathy syndrometype 1 (APS-1).179,180 APS-1 is remarkable because of the progressive development of multiple organ-specific autoimmune diseases, most frequently autoimmune adrenocortical failure and hypoparathyroidism, but also autoimmune thyroid disease, diabetes, vitiligo, alopecia, ovarian failure, hypogonadism, liver disease, pernicious anemia, and exocrine pancreatic insuffi-ciency.181 Almost all patients develop mucocutaneous Candida albicans infections because they produce neutralizing autoantibodies to IL-17, a cytokine that is essential for immune control of Candida.182,183 While this experiment of nature established that AIRE underpinned a critical mechanism for self-tolerance to a wide range of discrete organs, the AIRE gene and protein provided few clues to its mechanism of action: it contained motifs that suggested a role in transcription regulation,179,180 it was mostly expressed in the thymus and not in the organs that were affected by autoimmunity,180 and in the thymus the protein was primarily expressed in medullary thymic epithelial cells where it was concentrated in nuclear dots.184
mice could be rendered tolerant to adjuvant-free, deaggregated gammaglobulin from other species,6,33,34 and this tolerant state would persist for months provided the animals had been thymectomized to prevent generation of fresh T cells.198 However, unlike in the thymus, where the engagement of strong TCR-pMHC interactions leads to clonal deletion or anergy, strong pMHC signals in mature T cells normally leads to the development of an effective immune response. Therefore, with mature T cells we are left with the puzzle of how a strong TCR interaction results in tolerance versus activation. Solving this puzzle holds the most practical potential for overcoming the major clinical problems of transplant rejection and tumor immunology and for restoring tolerance in people with autoimmunity or allergy.
This antigen-tolerant state, dubbed T cell clonal anergy by Schwartz and coworkers, was not accompanied by long-lasting downregulation of surface CD3 and TCR. The missing costimulatory signal from the antigen-presenting cells could not be replaced by exogenous IL-1 or IL-2,232,233,234 but was later shown could be replaced by stimulatory antibodies against CD28.235 The tolerance-inducing signal from antigen on fixed antigen-presenting cells could be mimicked by selectively elevating intracellular calcium and required calcium-activation of calcineurin and induction of protein synthesis,233,234 leading to the concept that TCR-induced activation of calcium, calcineurin, and the transcription factor NFAT induced the expression of a lasting tolerance program in mature T cells if it was not accompanied by costimulatory signals.205,236
these studies was by Kyburz et al.,254 who compared the fate of TCR-transgenic CD8 cells recognizing a dominant peptide from LCMV presented by the MHC class I molecule, H2Db, after injecting thymectomized TCR-transgenic mice with LCMV peptide in incomplete Freund’s adjuvant (which provides no microbial costimulus but creates a reservoir of peptide). The specific CD8 T cells became large, increased in number, and developed cytotoxic T-lymphocyte (CTL) activity transiently, peaking on day 2 when many appeared apoptotic, with a rapid loss of blast cells and ex vivo CTL activity by day 3 and a drop in overall numbers below starting levels on day 5 and decreasing further on days 10 and 20. This abortive activation was independent of CD28, although CD28 deficiency made the response even more transient.256 The cells present after day 2 appeared functionally tolerant, as they could not be induced to proliferate or form CTLs in vitro from this time onwards, even when their subsequent deletion was abolished by transgenic expression of either Bcl-2 or Bcl-XL.255 Deletion of the activated T cells in this model thus resulted from activation of the intrinsic pathway of apoptosis, implying that the peptide-activated T-cell blasts received insufficient external signals for cell growth and metabolism, whereas the extrinsic apoptosis pathways triggered by Fas or TNFR1 had no measurable role.257,275
TABLE 32.1 Examples of Experimental Studies Demonstrating Abortive Proliferation, With or Without Anergy, Induced in Mature T Cells by Tolerogenic Antigens in Vivo | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|




