Immunologic Memory
Shane Crotty
Susan M. Kaech
Stephen P. Schoenberger
MEMORY AS A BIOLOGIC CONCEPT
The Protected State
Yet it was with those who had recovered from the disease that the sick and the dying found most compassion. These knew what it was from experience, and had now no fear for themselves; for the same man was never attacked twice- never at least fatally. And such persons not only received the congratulations of others, but themselves also, in the elation of the moment, half entertained the vain hope that they were for the future safe from any disease whatsoever.1
This quote comes from the Greek historian Thucydides (ca. 460 BC to ca. 395 BC) and describes his firsthand account of an ancient plague that struck Athens during a protracted war with Sparta. Although the identity of the causative agent continues to be a matter of speculation,2 these words transcend the ages to vividly illustrate the concept of immune memory—the phenomenon in which prior exposure to an infectious pathogen endows an individual with immunity, a durable state of protection against reinfection with the same organism. Unknown to Thucydides and his fellow Greeks, the immunity observed in the survivors of this plague reflected changes in the operational status of their immune systems as a result of the first (primary) response to the infecting pathogen. In the act of responding to the initial infection, the immune system expands a diverse population of antigen-specific B- and T-lymphocyte clones possessing a range of affinities and effector capacities. Through a process that is just now beginning to be understood at the molecular level, a portion of this repertoire is retained within the memory pool in the form of cells that are able to persist for long periods of time at relatively stable numbers by maintaining a slow but steady rate of division that is roughly equivalent to their rate of loss (death). When faced with a renewed challenge from the same (or antigenically related) infectious agents, memory cells mount a strong and rapid effector response that is capable of stopping reinfection at its earliest stages. The functional units of immune memory, therefore, are the long-lived B and T cells that mount rapid secondary (recall) responses upon reencounter with their cognate antigen. In the broader teleologic concept of memory, however, it is the antigen receptors that were shown to be “useful” in combating the initial infection that are selected and preserved at an increased frequency on clonal progeny that possess enhanced response kinetics and specialized functions. Whether generated through infection or vaccination, the value of the protected state they confer to the individual is self-evident, as preexisting immunity can prevent or limit the potential damage of an otherwise unrestrained infection. This can be of benefit from the earliest days of life, as seen in the example of maternal antibodies transmitted to the neonate in milk and serum that may serve to limit infections and transform them into “natural vaccinations” and thereby diminishing the severity of childhood infections that will be encountered during the next 1 to 2 years.3
This chapter will examine the phenomena and mechanisms of long-lasting protection that can develop following infection or vaccination, and will focus on the factors that govern the establishment and maintenance of specialized subsets of lymphocytes generated from the naïve repertoire during the primary response to a given antigen. This concept stands in contrast to the “memory” associated with immune tolerance, through which the adaptive arm of the immune system can prevent inappropriate responses against ubiquitous self-antigens. Immune memory involves a stable increase in the number of antigen-experienced B- and T-lymphocytes that have acquired specialized functional properties, allowing them to generate secondary responses that are more rapid and effective than those made by their clonal antecedents during the primary response. It is in the establishment, maintenance, and execution of memory responses that the adaptive immune system finds its greatest purpose for the preservation of both the individual and population. As will be discussed in this chapter, immune memory depends on a remarkable degree of interaction and cooperation between many different cell types in an elegant division of labor aimed at preserving those T- and B-cell specificities that have proved useful in the battles fought and won against previously encountered pathogens. It is important in considering the concept of immune memory, however, that distinctions be drawn between the protected state, an operational definition that can vary with the nature and magnitude of the antigenic challenge used to test its integrity, and memory cells, the clonal elements of memory whose contribution to the protected state can only be inferred from their phenotypic and functional properties.
Lifelong Memory can be Induced through Infection or Vaccination
Observations of acquired resistance to recurring diseases are recorded throughout the history of human epidemics. The careful observations of the Danish physician Ludwig Panum were among the first to clearly illustrate just how durable the protected state can be in an individual. Working on the
remote Faroe Islands in the North Sea, Panum studied two separate measles epidemics that took place 65 years apart between the 18th and 19th centuries. In the apparent absence of outbreaks in the intervening period, he found that people who had contracted measles in the first epidemic were not affected during the second, while those of appropriate age that were infected during the second outbreak had not been affected during the first.4 Thus, Panum observed, a single infection was capable of endowing an individual with lifelong immunity through a process that did not require reexposure to the pathogen. Although immune reactivity can be boosted by repeated exposure to a given organism, the extent to which this determines the maintenance of immune memory remains a source of some controversy.5 A source of uncertainty in applying this concept to human immunology is the difficulty in determining whether a single or multiple exposures have taken place before the memory state is assessed. With infrequently encountered life-threatening infections such measles, yellow fever, or polio, it is relatively straightforward to infer that a single exposure can establish a state of protection for up to 75 years that appears to be mediated by antibodies.4,6,7 With less threatening ubiquitous agents such as those causing the childhood infections chicken pox (Varicella zoster) and whooping cough (Bordetella pertussis), it is unclear whether immune memory may depend on occasional subclinical reinfection.8
remote Faroe Islands in the North Sea, Panum studied two separate measles epidemics that took place 65 years apart between the 18th and 19th centuries. In the apparent absence of outbreaks in the intervening period, he found that people who had contracted measles in the first epidemic were not affected during the second, while those of appropriate age that were infected during the second outbreak had not been affected during the first.4 Thus, Panum observed, a single infection was capable of endowing an individual with lifelong immunity through a process that did not require reexposure to the pathogen. Although immune reactivity can be boosted by repeated exposure to a given organism, the extent to which this determines the maintenance of immune memory remains a source of some controversy.5 A source of uncertainty in applying this concept to human immunology is the difficulty in determining whether a single or multiple exposures have taken place before the memory state is assessed. With infrequently encountered life-threatening infections such measles, yellow fever, or polio, it is relatively straightforward to infer that a single exposure can establish a state of protection for up to 75 years that appears to be mediated by antibodies.4,6,7 With less threatening ubiquitous agents such as those causing the childhood infections chicken pox (Varicella zoster) and whooping cough (Bordetella pertussis), it is unclear whether immune memory may depend on occasional subclinical reinfection.8
Given the long history of immunity in survivors of infectious disease, it is notable that even the most rudimentary understanding of how the protected state could be achieved was realized long after methods for its induction had been devised. Among the earliest practices was that of variolation, which had long been in practice in China and India as a means for enhancing protection against the deadly smallpox virus, which had been a major source of mortality in humans since recorded history.9 Variolation involves the intentional inoculation with desiccated material obtained from the open pustules of smallpox victims, in hopes that exposed individual would develop a milder form of the disease and, upon recovery, would be immune to smallpox (variola virus). Although effective in many cases, the procedure was not without risk to the recipient, who risked contracting a more serious life-threatening form of the disease that could be spread to others. In 1796, Edward Jenner famously (and dramatically) overcame these obstacles by using material obtained from the pustules of milkmaids infected with cowpox (vaccinia virus), an antigenically related but less virulent type of poxvirus, to inoculate young James Phipps who subsequently survived his exposure to infectious smallpox patients.10 In the 19th century, Louis Pasteur further developed the concept of using less-virulent or intentionally disabled forms of infectious organisms to confer immunity to bacterial diseases such as chicken cholera and anthrax, or viral diseases such as rabies. Pasteur used the word vaccine as a generic term to describe antigenic preparations administered to produce immunity, in reference to Jenner’s earlier work with cowpox (vaccinia) virus (vacca is Latin for cow). As described in more detail in the chapter on vaccines (Chapter 43), current vaccines can be generated from organisms that are antigenically related to the one against which immunity is desired, from inactivated forms of the entire disease-causing organism, or from specific constituent portions of the infectious organism such as proteins, polysaccharides, or nucleic acids.
Despite their various compositions, all effective vaccines recapitulate several important aspects of the immune response leading to memory formation: they mimic the threat of an infectious pathogen through stimulation of innate immunity, and they contain distinct antigens that can become recognizable by cells of the adaptive immune system.11,12 The power of vaccination to induce long-lasting immune memory is best illustrated by the example of the smallpox vaccine (vaccinia). This once-devastating organism has been virtually eradicated since the late 1970s through a worldwide mass vaccination program that still stands as one of the crowning achievements of the 20th century.13 The immunity induced by the smallpox vaccine is of remarkable durability, with both antibody and cluster of differentiation (CD)4+ and CD8+ T cell responses detectable 50 years after initial priming,14,15,16 and effectiveness, with a lifelong protection rate of 90% to 95% in vaccinated individuals, and a fatality rate of only 2% in those vaccinated within a decade before exposure.17,18 With the advent of modern immunologic tools such as enzyme-linked immunosorbent assay, intracellular cytokine staining, and enzyme-linked immunospot assays for the detection of rare vaccinia-specific T and B cells, a more detailed longitudinal picture of the response to the smallpox vaccine has emerged. These studies suggest that humoral immunity is more durable than cellular immunity, as increased numbers of vaccinia-specific memory B (Bmem) cells remain relatively stable for more than 50 years after vaccination, whereas CD4+ and CD8+ T-cell responses continue to decline over time, with a half-life of 8 to 15 years.14,16,19 This indicates that Bmem-cell responses, and to a lesser extent memory T-cell responses, are maintained by robust mechanisms.
Given the remarkable longevity of the immunity induced through either infection or vaccination, it is clear that the cells mediating the protected state must possess several key properties in order to preserve useful specificities generated in previous antigenic experiences, with the study of memory T cells revealing insights into a number of these features. First among these is that antigen-specific memory cells must be present in the immune repertoire in greater numbers than in the naïve repertoire as a result of their initial proliferation.20,21,22,23,24,25 Secondly, they must have undergone a program of differentiation involving alterations in chromatin structure and activation of specific transcription factors to allow the expression of key effector molecules such as cytokines, chemokines or cytotoxic proteins, in the case of cytotoxic T lymphocytes (CTLs), immediately upon recognition of antigen-expressing target cells.26,27,28,29,30,31,32,33 Memory cells must be able to survive as a clonal population over time; that is, they must be able to replace cells lost to normal homeostatic turnover.34,35 Memory cells also must perform a sentinel function by occupying specific physiologic niches where naïve cells are not usually found and where antigen exposure will either occur or be detected such as the mucosa of the lung or gastrointestinal tract.36,37 Consistent with this
sentinel function, memory cells are found to be capable of responding to lower concentrations of antigen than naïve cells through the selective expansion of high-affinity clones and the upregulation of adhesion molecules and reorganization of their T-cell receptor (TCR).38,39,40,41 Lastly, memory cells should be able to respond quickly to reencounter with their cognate antigen with the induction of rapid effector functions and renewed rounds of clonal expansion.42 Memory cells have developed a variety of mechanisms to achieve each of these specialized properties. In the following section, we will examine how memory cells are generated during immune responses and how these key features are imparted during priming, maintained over long periods of time, and expressed when needed to confer rapid protection from reinfection (Fig. 31.1).
sentinel function, memory cells are found to be capable of responding to lower concentrations of antigen than naïve cells through the selective expansion of high-affinity clones and the upregulation of adhesion molecules and reorganization of their T-cell receptor (TCR).38,39,40,41 Lastly, memory cells should be able to respond quickly to reencounter with their cognate antigen with the induction of rapid effector functions and renewed rounds of clonal expansion.42 Memory cells have developed a variety of mechanisms to achieve each of these specialized properties. In the following section, we will examine how memory cells are generated during immune responses and how these key features are imparted during priming, maintained over long periods of time, and expressed when needed to confer rapid protection from reinfection (Fig. 31.1).
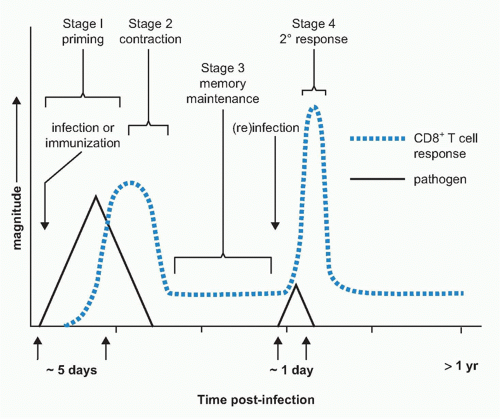 FIG. 31.1. Stages of the T-Cell Response to Infection. Antigen-specific T cells clonally expand during stage 1 and differentiate into effector T cells. After viral clearance, the majority of the cells die over the next 2 to 4 weeks (ie, stage 2, the bulk contraction phase). Soon thereafter, the number of virus-specific T cells stabilizes and the cells enter the memory phase and are maintained for long periods of time (stage 3). The majority of the memory T cells undergo slow, periodic basal homeostatic proliferation in response to interleukin-15 and interleukin-7. Upon reinfection, the memory T cells rapidly produce effector cytokines and other antimicrobial molecules and proliferate robustly to thwart infection. |
THE GENERATION OF MEMORY T CELLS
Introduction
Antigen-driven T-cell responses can be broadly categorized as having two main components: a comparatively shortlived effector arm that comprises large numbers of cells that mediate direct functional response against target cells, and a smaller but longer-lived memory population that insures the potential of rapid and potent recall responses against the inducing antigen. The memory component can itself be distinguished into two separate activities: one that resides mainly within lymph nodes where it can respond to antigenic challenge by renewed clonal expansion and production of secondary effectors (“central memory” phenotype T cells [TCM]) and one that is largely disseminated throughout peripheral tissues where the first contact with infectious pathogens is likely to occur (“effector memory” [TEM] or “resident memory” [TRM] phenotype T cells), which are capable of mediating immediate effector functions upon antigen recognition.36,42,43,44 Memory T cells disseminate widely throughout the body and circulate passing from blood to tissues and lymphoid organs, but more tissue resident TRM, noncirculating memory T cells, can also be found in the brain and mucosal tissues such as the gut and skin that reside long-term in the tissues following infection.45 The specific roles in immunity and the signals that may govern the development of these subsets will be discussed in subsequent sections in this chapter.
Models of Effector and Memory T-Cell Development
There are several models for how both effector and memory populations could be concomitantly generated, either in parallel or sequentially (Fig. 31.2). 1) The first model suggests that memory cells are the clonal progeny of a distinct precursor population that follows a separate differentiation fate from that of the primary effectors, and instead survive to execute these effector functions during the recall phase (separate precursor model). However, little support for this model exists currently because elegant lineage tracing experiments have demonstrated that the progeny of a single, naïve antigen-specific CD8 T cell can have diverse fates.46,47 2) The second, the “decreasing potential model,” postulates that increasing amounts or prolonged periods of TCR stimulation causes the effector T cells to progressively lose memory cell potential and, ultimately, reach a terminally differentiated state.48 This may occur in a linear or step-wise manner, driving the T cells through several distinct differentiation steps. Although this model was originally based mostly on antigenic signaling (signal 1), it is likely the collective history of signaling through costimulatory receptors (signal 2) and
inflammatory cytokines (signal 3) that determine the differentiation set-points of the T cells during infection. 3) The third model also allows for formation of heterogenous effector cell populations dependent on the overall “strength” of the signals 1, 2, and 3 encountered early during T-cell priming as first described by Lanzavecchia and Sallusto.49 High or excessively strong signals drive greater clonal expansion, but might also cause terminal effector T-cell differentiation. This model differs conceptually from the decreasing potential hypothesis in that different cell fates can be specified early according to the intensity of the signals received, but do not necessarily require multiple rounds of stimulation to create heterogeneous cell fates. 4) This model is also conceptually similar to an alternative “asymmetric cell fate model”
that supports the notion that TEM and TRM fates can arise from a single precursor T cell via asymmetric cell division that occur as the activated T cells clonally expand, and may even begin as early as the first cell division.50 This fourth model envisions memory as a cellular fate governed by the same common biologic strategy used during ontogeny and development to specify distinct functional properties in selected cells and their clonal progeny.51 The latter three models are not mutually exclusive, and elements of each of these models most likely occur simultaneously during an immune response. Moreover, although there is some experimental evidence for early cell fate commitment, a more likely scenario is that signals encountered early during T-cell priming setup or specify T-cell fates, but that some plasticity remains in the dividing cells and their differentiation states and fates become determined through the subsequent contact with antigen-presenting cells (APCs) or other signals over the next several days of infection.
inflammatory cytokines (signal 3) that determine the differentiation set-points of the T cells during infection. 3) The third model also allows for formation of heterogenous effector cell populations dependent on the overall “strength” of the signals 1, 2, and 3 encountered early during T-cell priming as first described by Lanzavecchia and Sallusto.49 High or excessively strong signals drive greater clonal expansion, but might also cause terminal effector T-cell differentiation. This model differs conceptually from the decreasing potential hypothesis in that different cell fates can be specified early according to the intensity of the signals received, but do not necessarily require multiple rounds of stimulation to create heterogeneous cell fates. 4) This model is also conceptually similar to an alternative “asymmetric cell fate model”
that supports the notion that TEM and TRM fates can arise from a single precursor T cell via asymmetric cell division that occur as the activated T cells clonally expand, and may even begin as early as the first cell division.50 This fourth model envisions memory as a cellular fate governed by the same common biologic strategy used during ontogeny and development to specify distinct functional properties in selected cells and their clonal progeny.51 The latter three models are not mutually exclusive, and elements of each of these models most likely occur simultaneously during an immune response. Moreover, although there is some experimental evidence for early cell fate commitment, a more likely scenario is that signals encountered early during T-cell priming setup or specify T-cell fates, but that some plasticity remains in the dividing cells and their differentiation states and fates become determined through the subsequent contact with antigen-presenting cells (APCs) or other signals over the next several days of infection.
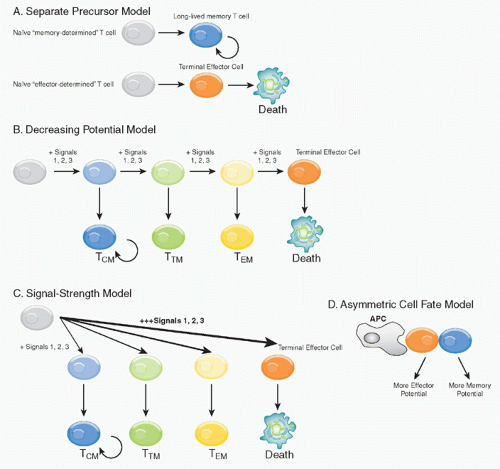 FIG. 31.2. Models of Memory T-Cell Generation. Four models are proposed to describe the formation the memory T cells. Please see text for detailed descriptions of each model. A: Separate precursor model. This model suggests that naïve T cells can be preprogrammed following thymic development to develop into memory T cells or terminal effector cells that die following infection. B: Decreasing potential model. This model states that a short duration of antigenic stimulation favors development of effector cells that will give rise to greater numbers of central memory phenotype T cells (TCM; blue), whereas longer durations of stimulation promotes terminal effector cell differentiation (red) and death. According to the number of interactions with antigen-presenting cells and possibly cell divisions, intermediate stages of effector differentiation can also be acquired, giving rise to memory T cells with more effector memory phenotype T cell (TEM)-like (orange) or transitional memory (TTM; yellow) phenotypes that can convert from TEM → TCM phenotypes overtime. Curved arrow denotes basal homeostatic proliferation. C: Signal strength model. This model proposes that the effector cells differentiate according to the “strength of signal” they receive at the time of priming (or possibly later). Larger amounts of antigen, costimulation, and inflammation (thicker arrow) drive cells toward a more terminal differentiation state, whereas weaker signals (thinner arrows) promote formation of TCM, TTM, or TEM cells in a progressive manner. D: Asymmetric cell fate model. This model proposes that T-cell fates are specified (or possibly even determined) as early as the first cell division by the asymmetric portioning of cell fate determinants. |
In each of these models, the initiating event leading to memory cell development is the primary activation of a naïve T cell in response to immunization or infection. We shall therefore begin our consideration of memory T-cell generation within the different phases of the immune response: 1) the first phase of naïve T-cell priming, clonal expansion, and effector cell differentiation (the naïve to effector T-cell transition); 2) the second phase of effector cell contraction and formation of a surviving pool of memory T cells (the effector to memory T cell transition); 3) the third phase of memory T-cell maintenance and homeostasis; and 4) the reactivation of memory T cells during reinfection. Within each phase, we will discuss the similarities and differences between CD4 and CD8 T cells in effector and memory T-cell development.
Methods of Detecting Effector and Memory T Cells
Before discussing the factors responsible for the development of effector and memory T cells, it is important to understand the types of experimental approaches that have been used in their study and how these influence the various conclusions that can be drawn from the experimental data. In general, there have been two main experimental settings through which memory T-cells have been studied in vivo. One involves enumeration of cells that express markers associated with antigen-experience such as CD44hi or CD62Llo for activated cells, in either immunized or nonmanipulated normal mice. In an immunized host, the antigen-specific T cells can be generated from the endogenous polyclonal repertoire or adoptively transferred monoclonal transgenic T cells. In the latter case (a naïve host), the cells in question are the spontaneously arising “memory phenotype” (MP) T cells of unknown derivation and specificity, which increase in frequency with age.52,53 In contrast, antigenspecific effector and memory T cells can be distinguished and tracked longitudinally in mice and humans using major histocompatibility complex (MHC) class I or II tetramers, which are soluble forms of the ligand structure recognized by T cells and allow the detection of cells recognizing a specific antigenic determinant.54,55 Alternatively, antigen-specific T cells can also be identified functionally using intracellular cytokine staining, which allows enumeration of cells secreting cytokines in response to their cognate peptide antigen. Finally, the use of TCR-transgenic T cells has allowed the fate of a monoclonal population of antigen-specific T cells to be followed throughout their development from naive to memory cells during immune responses in mice. This typically involves the adoptive transfer of relatively small numbers of naïve TCR-transgenic T cells into a wild-type mouse that is subsequently immunized or infected to activate the T cells.
Phase I. Naïve to Effector T-Cell Transition: T-Cell Priming, Clonal Expansion, and Effector Differentiation
T-Cell Priming
The process of generating T-cell memory begins at priming, which is the initial activation of mature postthymic precursors (ie, naïve T cells) by foreign antigens presented by professional APCs. This is not a simple task, as a T cell recognizing any one single antigen is rare in the naïve repertoire, occurring with a frequency of perhaps 1 in 105 to 106 among the total population.56,57 The antigen-bearing APCs are also likely to be infrequent among the (tens of) millions of cells within a given lymph node or in the spleen, as in most cases they must first obtain their antigenic cargo at peripheral sites of infection and then migrate via the lymphatics to lymph node or via the bloodstream to the spleen.58 As will be described, the complex interaction of rare and migrating cell types necessary to the generation of immune responses is facilitated by highly regulated patterns of migration and by the structural features of the secondary lymphoid organs themselves.
Naïve T cells are produced at a modest rate by the thymus, with approximately 1 to 2 × 106 cells per day emerging into the bloodstream in young mice.59,60 The survival of naïve cells/recent thymic emigrants requires at least two types of extrinsic signals: 1) MHC molecules that are presumably occupied with low-affinity ligands (self- or environmental peptides) and provide tonic TCR stimulation, and 2) a critical cytokine, interleukin (IL)-7, which has many roles in B- and T-cell lymphopoeisis.61,62,63,64,65,66,67,68 Mice or humans deficient in either IL-7 or IL-7Rα have considerably reduced numbers of T cells.69,70,71,72,73,74 IL-7 is a member of the common γ-chain (γc) cytokine family that signals through a heterodimer comprised of γc and IL-7Rα.75 Neither of these components are unique to IL-7, as γc is shared with IL-2, IL-4, IL9, IL-15, and IL-21, and IL7Rα is shared with thymic stromal lymphopoietin (TSLP).
To become stimulated (primed), naïve T cells must recognize antigens in the form of peptide fragments bound to MHC molecules on professional APCs, such as dendritic cells (DCs), in lymphoid organs. In response to microbial pathogens, immature DC migrate to sites of infection and inflammation in response to a range of CC and CXC chemokines and acquire soluble and particulate antigens. The immature DCs become activated by recognition of pathogen-associated molecular patterns or host molecules released by tissue damage and transition into an APC by
increasing expression of MHC:peptide complexes, the chemokine receptor CCR7 to facilitate migration toward T-cell areas of secondary lymphoid organs, and costimulatory ligands and cytokines to aide in T-cell activation and effector cell differentiation.76 Once a naïve T cell recognizes its cognate peptide/MHC ligand on an activated DC within the secondary lymphoid organs, the interactions of priming (ie, the “antigenic signal”) can take place. The molecular details of this process involve the formation of immune synapses at the T/APC contact site that are composed of rapidly-clustering of TCR molecules binding peptide/MHC complexes on APC plus the local accumulation of intracellular signaling molecules such PKC-θ, LAT, and LCK.
increasing expression of MHC:peptide complexes, the chemokine receptor CCR7 to facilitate migration toward T-cell areas of secondary lymphoid organs, and costimulatory ligands and cytokines to aide in T-cell activation and effector cell differentiation.76 Once a naïve T cell recognizes its cognate peptide/MHC ligand on an activated DC within the secondary lymphoid organs, the interactions of priming (ie, the “antigenic signal”) can take place. The molecular details of this process involve the formation of immune synapses at the T/APC contact site that are composed of rapidly-clustering of TCR molecules binding peptide/MHC complexes on APC plus the local accumulation of intracellular signaling molecules such PKC-θ, LAT, and LCK.
Activation of T Cells by Antigen (Signal 1) and Costimulatory Receptors (Signal 2)
Another critical consequence of DC activation is the upregulated expression of numerous costimulatory molecules and cytokines that contribute to T-cell priming by promoting cell division, survival, and differentiation.58,77,78,79 The primary activation of naïve T cells has traditionally been believed to require at least two signals: an antigen-specific “signal 1” that is transmitted upon TCR-mediated recognition of its cognate peptide/MHC complex, and an antigen nonspecific “signal 2” costimulus.80,81 This “two-signal” model predicted that engagement of the TCR in the absence of costimulation is insufficient for sustained clonal expansion and cytokine production, and cells instead become anergic and unresponsive to further stimulation.82 While this outcome has been demonstrated in a number of studies, the number of ligand/receptor pairs capable of functioning as “signal(s) two” has grown considerably in the last decade. CD80 and CD86 are considered to be the prototypical costimulatory molecules expressed on APC capable of transmitting “signal 2” through their interaction with CD28 expressed on T cells. CD80/86-mediated ligation of CD28 produces signals in T cells that are distinct from those of the TCR and enhances clonal expansion, effector functions, and memory development.83,84,85,86 Although many CD4+ and CD8+ T-cell responses depend on CD28, the requirement is not absolute, as many types of T-cell responses can occur in its absence in vivo, including allograft rejection, induction of allogeneic graft-versus-host and delayed-type hypersensitivity responses, and the generation of cytotoxic CD8+ T cells.87,88,89,90,91 These observations suggest that additional pathways exist for costimulation of T cells other than CD28.92 A number of these have emerged from the tumor necrosis factor receptor (TNFR) family (CD27/CD70, 4-1BB (CD137)/4-1BBL, OX40 (CD134)/OX40-L, HVEM-LIGHT, CD30/CD30-L, and GITR), each of which can provide costimulatory stimuli that synergize with TCR signals to sustain T-cell activation after priming, and that have been shown to be important for the magnitude and longevity of CD8+ T-cell responses in vivo in studies of knockout mice lacking either ligand or receptor.92,93,94 Interestingly, the costimulatory requirements for CD4 and CD8 T cells are not completely overlapping. For example, CD8+ T-cell activation and clonal expansion occurs efficiently in mice lacking CD28, CD40L, or OX40, whereas CD4+ T-cell responses are substantially reduced. The opposite situation is seen in mice lacking 4-1BB-/-, which mount normal CD4+, but diminished CD8+, T-cell responses to lymphocytic choriomeningitis virus (LCMV) infection.95 It should be pointed out that the influence of costimulatory interactions on the regulation of CD8+ T-cell responses is by no means limited to the priming event, as several receptor-ligand pairs, notably 4-1BB/4-1BBL, OX4O/OX40L, BTLA/HVEM, and PD-1/PDL1/PDL1, can act on the expanded population, with either positive or negative effects on T-cell function and survival.94,96,97
Inflammatory Cytokines (Signal 3) and Effector T-Cell Specification
In addition to contact-dependent costimulation, a number of inflammatory cytokines including interferon (IFN)α, IFNβ, IFNγ, IL-1, IL-4, IL-6, IL-12, and IL-21 have been implicated in the transmission of a “signal 3” that is required for optimal proliferation and differentiation of effector T cells.98,99 Signal 3 cytokines are produced mostly by innate immune cells in response to pattern recognition receptor activation, but the types of cytokines produced will vary according to the pathogen, its tropism, and the types of pattern recognition receptors and sentinel cells activated during infection or vaccination. This selectivity of cytokine production ensures development of the appropriate types of effector T cells to defend against the given infecting pathogen.99 For instance, intracellular pathogens, such as viruses and certain bacteria, elicit production of IFNα/β, IL-12, and IFNγ, which drive development of type I immune responses that mainly consist of cell-mediated defenses aimed at direct killing of infected cells via CD8+ CTLs or suppression of viral replication and activation of macrophages through TH1-polarized CD4+ responses.100 Infection by extracellular pathogens including parasites and helminthes produces TSLP and IL-4, which induces development of more humoral-based type 2 responses that generate Th2-polarized CD4+ effector T cells and recruitment of eosinophils, basophils, and mast cells. Other extracellular pathogens, including gram-negative bacteria, fungi, and some protozoa, induce IL-6 and IL-1; in the presence of basal transforming growth factor (TGF)-β, this instructs development of Th17 CD4+ T cells that recruit neutrophils and macrophages to sites of infection.101,102,103,104 Additionally, as discussed in greater detail subsequently, CD4 T cells also form follicular helper T cells,which are specialized for B-cell help and control of germinal center (GC) responses and antibody production.105 Lastly, regulatory T (Treg) cell populations that develop in the thymus or in the periphery prevent autoimmunity and, during infection, keep effector T cells in check to reduce the amount of collateral damage (ie, tissue damage) associated with immune responses. Additional varieties of CD4 T cells have been proposed that differ based on cytokine production or localization.106,107
Much progress has been made in elucidating the genetic programs that govern development of CD8+ CTLs and CD4+ TH1, TH2, and Th17 effector T cells. During type I responses (eg, antiviral responses) TCR and IL-12, IFNαβ, and IFNγ signaling act together to activate transcription factors such as signal transducer and activator of transcription factor (STAT) 4 and T-bet in CD4+ T cells to induce formation of IFNγ-producing TH1 cells. Likewise, in CD8+
T cells STAT4, T-bet and eomesodermin expression are needed to generate CTLs to directly kill infected cells.107,108,109,110 Similarly, during type 2 responses, IL-4-dependent activation of STAT6 and GATA-3 creates TH2 effector cells that produce IL-4, IL-5, and IL-13 as “signature cytokines” to trigger immunoglobulin (Ig)E production by B cells and activate eosinophils, basophils, and mast cells.107 This, for example, creates a “weep and sweep” mode of defense to combat infection of helminths.111,112,113 Although the exact details of TH17 development remain to be elucidated, the cytokines IL-6, IL-1, and TGF-β play an instructive role in their differentiation through engagement of STAT-3 and the nuclear orphan receptor RORγ.114,115,116,117,118 The development of TH17 or Treg cells appears to be reciprocal and dependent on the ratio of IL6:TGF-β as well the abundance of vitamin A metabolite retinoic acid.119,120,121 For example, steady-state production of TGF-β may favor Treg formation, but upon bacterial infection, the combination of newly synthesized IL-1 and IL-6 will favor Th17 differentiation.122 Signals for Tfh cell differentiation are incompletely characterized, but IL-6 and ICOS are important factors in most cases.105,123,124,125,126 In addition, Tfh cells depend on multiple stages of APC interactions, first requiring signals from DCs and then subsequent interactions and signals from B cells.125,127,128,129
T cells STAT4, T-bet and eomesodermin expression are needed to generate CTLs to directly kill infected cells.107,108,109,110 Similarly, during type 2 responses, IL-4-dependent activation of STAT6 and GATA-3 creates TH2 effector cells that produce IL-4, IL-5, and IL-13 as “signature cytokines” to trigger immunoglobulin (Ig)E production by B cells and activate eosinophils, basophils, and mast cells.107 This, for example, creates a “weep and sweep” mode of defense to combat infection of helminths.111,112,113 Although the exact details of TH17 development remain to be elucidated, the cytokines IL-6, IL-1, and TGF-β play an instructive role in their differentiation through engagement of STAT-3 and the nuclear orphan receptor RORγ.114,115,116,117,118 The development of TH17 or Treg cells appears to be reciprocal and dependent on the ratio of IL6:TGF-β as well the abundance of vitamin A metabolite retinoic acid.119,120,121 For example, steady-state production of TGF-β may favor Treg formation, but upon bacterial infection, the combination of newly synthesized IL-1 and IL-6 will favor Th17 differentiation.122 Signals for Tfh cell differentiation are incompletely characterized, but IL-6 and ICOS are important factors in most cases.105,123,124,125,126 In addition, Tfh cells depend on multiple stages of APC interactions, first requiring signals from DCs and then subsequent interactions and signals from B cells.125,127,128,129
Clonal Expansion of Effector T cells
The kinetics of T-cell clonal expansion can be remarkable. In murine model systems of Listeria and LCMV infection, it has been documented that CD8 T cells undergo between 15 to 20 sequential rounds of division within the first 7 days of infection, at rates as fast as ˜4 to 8 hours, and increase the frequency of antigen-specific T cells more than 10,000-fold over their initial frequency in the naïve.32,130,131,132,133,134,135,136,137 CD4 T cells also undergo profound clonal expansion, but their burst sizes are typically smaller than that of CD8 T cells. The factors that govern the magnitude of T-cell proliferation in vivo are numerous, but several studies have demonstrated a direct relationship between antigen load and the number of effector cells generated.135,136,138,139,140,141,142 However, it is important to point out antigen-driven T-cell proliferation cannot be sustained indefinitely, as in the case of chronic infections (such as human immunodeficiency virus [HIV] and hepatitis C virus infection in humans and LCMV infection in mice), where antigen persists for prolonged periods. That is, despite the presence of antigen, the antigen-specific T cells still undergo contraction 1 to 2 weeks post infection. In these situations, it has been postulated that this occurs due to “programmed” contraction of activated CD8 T cells, but another critical element is that these cells differentiate into senescent, less functional “exhausted” T cells that may be sensitive to apoptosis.143,144,145,146 Another difference observed between CD4+ and CD8+ T cells is that CD8+ T cells require a comparatively shorter period of stimulation (2 to 4 hours) for commitment to clonal expansion and effector development than do CD4+ T cells (˜20 hours).135,137,147,148 CD8+ T cells also begin dividing earlier after priming than CD4+ T cells and have a faster rate of division.148,149,150,151 Additionally, and in contrast to CD8+ T cells, it has been observed that CD4+ T cells require continued antigenic stimulation throughout their period of expansion in order to achieve full proliferative and effector potential.152,153 Based on these and other findings, Lanzavecchia and Sallusto have suggested that the optimal development of memory T cells will occur in cells that receive a specific level of antigenic stimulation during their priming and subsequent expansion that enables them to utilize and access survival signals in a process termed progressive differentiation.49,147,154,155
In addition to the effects of the inflammatory cytokines on effector T-cell specification, some of these cytokines also play a critical role in effector cell survival and clonal expansion. For instance, studies on CD8+ T cells primed in vitro with peptide/MHC and CD80 showed that optimal expansion and development of lytic functions required addition of IL-12 or IFNα.156 IFNαβR-deficient LCMV GPspecific CD8+ T cells transferred to wild-type mice showed dramatic reductions in their ability to undergo clonal expansion and to generate a memory pool following infection with LCMV.157,158 When the LCMV GP was expressed from a recombinant vaccinia virus, however, only a modest defect in CD8+ T-cell expansion and survival was observed, suggesting that the IFNαβ-dependence of CD8+ T-cell memory is influenced not by the target antigen, but rather by the immunogen.157,159 This idea has found support in a recent study showing that the response of IFNαβR-deficient transgenic CD8+ T cells to the same peptide-pulsed DC vaccine offered in the context of four different infections (LCMV, vaccinia virus, vesicular stomatitis virus, and Listeria monocytogenes) were most severely inhibited for LCMV infection.158,160 IFNγ has also been shown to support optimal CD8+ T-cell expansion after LCMV infection or peptide vaccination, and transgenic CD8+ T cells lacking the IFNγR1 display reduced expansion compared to wildtype cells in the same host.161,162,163 Thus, multiple types of innate-immune induced cytokines can boost expansion of activated T cells.
CD4+ T Cells Help CD8+ T-Cell Priming
Although DCs increase their immunogenicity in response to inflammatory stimuli delivered at peripheral sites of antigen uptake, their stimulatory capacity can be further enhanced within the lymph node through the action of CD4+ “helper” cells (TH) that recognize peptide antigens bound to MHC class II molecules at the surface of the same APC presenting to the CD8+ T cell164,165,166 (Fig. 31.3). The contribution of TH to CD8+ priming had initially been thought to be conditional (ie, required for response to some immunogens but not others) and limited in mechanism to the production of paracrine IL-2 for the benefit of CD8+ T cells present in a “three-cell cluster” together with APCs.167,168,169 More recent evidence, however, has shown that CD4+ T cells make important contributions to the generation and maintenance of CD8+ T cell responses at a number of discrete steps.165,170 CD4 T cells can produce IL-21 and IL-4, in addition to IL-2, to support CTL expansion.171,172,173,174,175 Several studies have shown that a key function of TH is that it involves the engagement of CD40 on DCs that leads to a number of immunostimulatory alterations, including high-level expression of
costimulatory and adhesion molecules, among them CD80, CD86, 4-1BB, OX40, and CD70, as well as high-level production of IL-12.94,176,177,178,179,180 CD4+ T cells can also help guide naïve CCR5-expressing CD8+ T cell precursors to relevant APCs within secondary lymph nodes by enhancing production of the chemokines CCL3 and CCL4 at the CD4+ T-cell/DC interface.181,182
costimulatory and adhesion molecules, among them CD80, CD86, 4-1BB, OX40, and CD70, as well as high-level production of IL-12.94,176,177,178,179,180 CD4+ T cells can also help guide naïve CCR5-expressing CD8+ T cell precursors to relevant APCs within secondary lymph nodes by enhancing production of the chemokines CCL3 and CCL4 at the CD4+ T-cell/DC interface.181,182
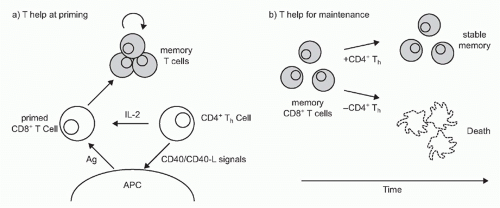 FIG. 31.3. Models of CD4+ T-Cell Help for CD8+ Memory Formation. CD4 T cells can help at two different stages of effector and memory CD8 T-cell development. A: CD4 T cells can help CD8 T-cell activation via multiple putative mechanisms including CD40:CD40L interactions between CD4 T cells and antigen-presenting cells (APCs), interleukin-2 secretion, or enhancing chemokine production by causing APCs. B: CD4 T cells also help generate functional memory CD8 T cells that are maintained long-term. |
The absence of TH can have different effects on the CD8+ T-cell response depending on the immunogen and the functional parameter measured. In certain settings of noninfectious immunizations using heat-killed or fixed preparations of pathogens or cocktails of foreign protein plus adjuvants, the extent of antigenic stimulation and inflammation can be relatively weak compared to immunization with live, replicating pathogens. In these cases, the antigens will often be cross-presented to CD8 T cells and their response will be CD4 T-cell dependent. In contrast, the primary CD8 T-cell response to many, but not all, acute viral or bacteria responses are only modestly decreased in the absence of CD4 T cells. It is thought that the overt stimulation of the innate immune response, and perhaps, more prolonged and widespread inflammation and antigen presentation delivers a stronger signal to the CD8 T cells to overcome the need of CD4 T cells for activation during infection. However, in chronic viral infections, where antigen persists, CD4 T-cell help becomes essential to sustain virus-specific CD8 T-cell survival and function overtime.183,184,185,186,187,188,189
Effector T-Cell Migration
Following commitment to clonal expansion and acquisition of effector functions, CD8+ T cells begin to express new surface patterns of chemokine, homing, and adhesion receptors that mediate their egress from lymph node and facilitate their migration to peripheral sites. An initial event in this process is the downregulation of CD62L and CCR7, the receptors used by naïve precursors to enter the lymph node, and restoration of the chemotactic response to the lysophospholipid sphingosine-1-phosphate through reexpression of its receptor (sphingosine-1-phosphate-receptor-1), which promotes migration to blood and lymph by distinct sources of its ligand.36,190,191,192 Following this, the migration of effector CD8+ T cells to nonlymphoid tissues will be largely determined by homing receptors and chemokines.193 These include chemokine receptors such as CCR2 and CCR5 that guide cells to inflammatory sites through recognition of a variety of chemokines induced by inflammatory cytokines. In some cases, and for reasons that are poorly understood, a subset of these cells will reexpress CD62L and CCR7, thereby allowing their reentry into the lymph node where they can function as TCM, the functional properties of that will be described in the following section.194 Others will migrate to the spleen via the blood and take up residence in the red pulp and are largely excluded from the white pulp.195 The ultimate “address” to which the cell will migrate, however, can be influenced by the conditions under which it was activated. For instance, in the intestine, DC from Peyer patches—specialized secondary lymphoid organs within the intestine—have been shown to induce the selective expression of the integrin α4β7and CCR9 chemokine receptor, both of which are essential for gut homing, in CD8+ T cells.196,197,198,199 Likewise, epicutaneous priming of T cells enhances expression of E-and P-selectin ligands and their subsequent homing to the skin via chemokine receptors CCR4 and CCR10, which bind CCL17 expressed by skin vascular epithelium and TACK expressed on keratinocytes.200,201,202 Thus, DCs not only “alert” naïve T cells to the presence of an infectious pathogen, but also instruct T cells to express the appropriate homing
receptors that enable their migration to the specific anatomic site where they are needed.37
receptors that enable their migration to the specific anatomic site where they are needed.37
Phase II: Effector to Memory Transition: Resolution of Immune Response and Formation of Memory T Cells
Contraction of Effector T-Cell Populations
Coincident with pathogen clearance at the end of the primary response, the majority of effector T cells are eliminated through a complex and poorly understood process that involves their orderly apoptosis (programmed cell death) via interactions between growth factors, various member of the TNFR superfamily and elements of the mitochondrial pathway of apoptosis.203,204 The contraction phase leaves a small population of cells, ˜5% to 10% of the numbers present at the peak of the response, to survive as memory cells, which in some cases remain stable for the life of the animal.205,206 The removal of effector cells achieves two important goals for the immune system: it prevents the T-cell repertoire from being overly populated or “focused” with T cells specific to a particular pathogen, and it restores homeostasis to the available environmental niches for support of T cells so that new antigenic challenges can be met. The magnitude of expansion of different epitope-specific T-cell populations can vary during an infection, creating what is referred to as “immunodominant” or “subdominant” T-cell populations based on their relative cell numbers. Interestingly, for CD8 T cells, the magnitude of contraction remains fairly constant across both immunodominant and subdominant clones such that a similar frequency of cells (˜90% to 95%) die during the contraction phase.21,131,132 These data suggest that the process of contraction occurs fairly equally across various populations of effector T-cell clones, irrespective of differences in precursor frequency and/or the number of times the cells may have divided during clonal expansion.
The loss of antigen-specific T cells at the end of immune responses is largely attributable to apoptotic death, a process traditionally characterized by the activation of a caspase cascade that results in the death of the host cell by cleavage of critical cellular proteins and creation of deoxyribonucleic acid double-stranded breaks.207 There are two main pathways by which caspases can be activated in T cells: the death receptor pathway and the mitochondrial pathway.206,208,209 Although the regulation of these pathways in activated T cells in vivo is not entirely clear, several chief factors are thought to be involved: 1) The abundance and duration of antigen is a major determinant of clonal burst size of activated T cells, and stemming from the original observation that restimulation of activated T cells with antigen in vitro can induce cell death, a process known as activation-induced cell death (AICD), antigenic stimulation likely also modulates T-cell death during infection.210 For example, in certain cases of chronic viral infection, such as LCMV infection in mice, it is thought that persistent antigenic stimulation can lead to the physical deletion of certain epitope-specific CD8 T-cell populations, leaving holes in the repertoire of pathogen-specific T cells.187,189 However, this does not happen to all virus-specific CD8 T cells during chronic LCMV infection, as virus-specific CD8 T cells directed against other viral epitopes are spared but develop into less functional CTLs, referred to as “exhausted” T cells that express inhibitory receptors that reduce TCR signal transduction.146,189 In contrast, during other latent infections, such as the murine cytomegalovirus (a herpes virus), some epitope-specific populations contract like normal, but others recognizing latently produced viral proteins may actually “inflate” after the acute phase of infection due to low-grade antigen persistence from viral reactivation.211,212 2) Likewise, subsequent encounter with death receptor ligands during or near the end of their clonal expansion phase can also affect effector cell contraction.213,214,215 3) The third is the decline in T-cell growth factors and survival signals, such as IL-2, following resolution of infection. In this scenario, it is postulated that the effector cells compete for the dwindling supply of resources. 4) The fourth is the intrinsic heterogeneity of the effector cell population and the balance of proapoptotic (Bim, FADD, FLICE, etc.) and antiapoptotic (c-FLIP, Bcl-2, Bcl-XL, etc.) proteins they express.207,216 5) Lastly, the anatomic location of the T cells within tissues may also regulate the exposure of effector and memory T cells to signals that promote survival or apoptosis.34,35,36,217,218,219 In the following, we will outline the major molecules that have been found to control effector T-cell apoptosis following infection.
Extrinsic Control of Effector T-Cell Apoptosis. The death receptor pathway activates apoptosis via the interaction of TNFR family receptors with protein activators such as TNFα and effector caspases 3 and 7.208,220 With regard to the death receptor pathways, fewer effector cells died following viral infection in mice lacking both p55 and p75 TNFRs, but surprisingly, there was little effect of deleting Fas or Trail.213,214,221,222,223,224 This result was unexpected given the critical role of Fas in T-cell homeostasis and the in vitro process of AICD.210,225 However, Fas was found to play an important role in contraction of antigen-specific T cells during chronic infections or autoimmune situations where prolonged interaction with antigen occurs.215,226,227 Moreover, Trail also plays a more prominent role in controlling the survival of memory CD8 T cells that are formed without robust CD4 T-cell help.224,228,229 Thus, control of effector and memory T-cell survival operates differently according to the type of situation encountered.
In addition to the death-inducing signals delivered by some TNFR family members, others also serve a role in costimulating T cells during priming, as discussed previously, and inducing survival signals for activated CD8 T cells during the contraction phase. In particular, interference with or deletion of 4-1BB or OX40 on T cells impairs effector cell expansion and augments their contraction following viral infection. Consequently, fewer memory T cells develop in the absence of these receptors.230,231,232,233 Interestingly, 41BB preferentially functions in CD8+ T cells and OX40 preferentially functions in CD4+ T cells, albeit OX40 also has a role in promoting effector CD8 T-cell differentiation and survival.234,235 Additionally, the cytokines IL-7 and IL-15 upregulate the costimulatory TNFR family members, OX40 and 4-1BB, respectively, and this may provide additional survival signals to activated T cells.236,237 Thus, distinct
niches may be created between DCs and antigen-specific T cells during the contraction phase whereby the interactions between cytokines, cytokine receptors, and TNFRs sustain T-cell survival.236
niches may be created between DCs and antigen-specific T cells during the contraction phase whereby the interactions between cytokines, cytokine receptors, and TNFRs sustain T-cell survival.236
Intrinsic Control of Effector T-Cell Apoptosis. Another form of T-cell apoptosis referred to as activated T cell autonomous death (ACAD) differs from the death receptor pathway in that it does not initiate from a specific receptor, but rather from disruption of the mitochondrial or lysosomal membranes, leading to activation of proteases such as caspases or cathepsins, respectively.238 The mitochondrial-regulated pathway of apoptosis is well characterized and initiates upon disruption of the mitochondrial membrane and release of cytochrome c into the cytoplasm. This integrates into a multisubunit complex (the apoptosome) to activate the downstream effector caspases 3 and 7,205,208 an endpoint common to the death receptor pathways mentioned previously.206,239 ACAD also involves the proapoptotic activities of the Bcl-2-related protein Bim and PUMA (p53-upregulated modulator of apoptosis) that are induced by a variety of cellular stressors including the withdrawal of key survival cytokines such as IL-2, IL-7, or IL-15.204,221,240,241,242,243,244,245 BIM can bind Bcl-2, BCL-XL, and MCL-1 on the mitochondrial membrane, and promote the release of cytochrome c and the induction of cell death.246,247 During their expansion phase, T cells also decrease their expression levels of Bcl-2 and Bcl-XL and thereby allow for activation of the intrinsic ACAD pathway.248 Indeed, ACAD, rather than AICD, is thought to be responsible for the contraction of CD8+ T cells that follows the expansion phase because a single deficiency in Bcl-2 results in greater death of effector T cells, and conversely, single deficiency in Bim or double deficiency in Bax and Bak profoundly enhances effector T-cell survival following infection.215,227,242,249,250,251,252,253,254 Interestingly though, Bim-/- T cells do gradually decay, approaching near wild-type numbers several months after infection.215,227,251,252 This finding suggests that alternative homeostatic control mechanisms and/or possibly competition for survival niches or factors, ultimately control T-cell numbers in the face of Bim deficiency.
The actions of effector T cells themselves, especially cytotoxic T cells, can cause self-inflicted injury and promote cell death. For instance, effector molecules such as perforin, granzymes, and IFNγ play a role in contraction, as mice deficient in either of these have increased numbers of CD8+ T cells in both the expansion and contraction phases.161 A set of proteins involved in protecting CTLs from their own cytotoxic machinery are the serine protease inhibitors Spi2a and Spi6.255,256,257 Spi2a inhibits the activation of cathepsin B in the lysosomal cell death pathway, a lesser-characterized cell death program that may involve both caspase-independent and -dependent mechanisms.257 When overexpressed, Spi2a was cytoprotective and reduced effector CD8 T-cell contraction following LCMV infection. In contrast, “knockdown” of Spi2a increased the magnitude of effector cell death and fewer memory CD8 T cells formed. Another serine protease inhibitor, Spi6, that targets granzyme B, maintains the integrity of cytotoxic granules by inhibiting the release of granzymes into the cytosol and preventing CTL “suicide.”256,258 Consequently, Spi6-deficient CD8 T cells have substantially reduced clonal expansion during viral infection.256
Heterogeneity of Effector T Cells and Memory Cell Potential
The previous section described numerous mechanisms that determine how effector T cells die following clonal expansion, but the factors that actually determine which of the effector cells die or live to populate the memory T-cell pool remains to be better resolved. Traditionally, it was proposed that effector T-cell death was more or less random and dependent on the competition between cells for limiting T-cell survival factors, such as common-gamma chain cytokines (IL-7, IL-2, IL-15). This permissive model implied that all the effector cells were equipotent in their ability to persist and give rise to memory T cells, and it was availability of extrinsic signals that ultimately determined their fate. Although competition for survival signals in cellular niches does impact effector and memory T-cell homeostasis, it is also evident that the effector cell population is heterogenous and contains effector cells that are intrinsically more “fit” to survive long-term and develop into memory T cells. These cells have been referred to as memory precursor cells (MPCs) and have best been characterized in CD8 T-cell populations based on IL-7Rα expression following acute viral and bacterial infections. Both naïve and memory CD8+ T cells express high levels of IL-7Rα, but this receptor is downregulated in the majority of effector CTLs during clonal expansion.64,70,259,260,261 However, a small percentage of the effector cells express relatively higher amounts of IL-7Rα and separation of virus-specific IL-7Rαhi and IL-7Rαlo CD8 T cells from LCMV-infected animals demonstrated that IL-7Rαhi effector cells had the greatest capacity to give rise to long-lived memory CD8 T cells that could proliferate and protect against secondary infection.260,262,263 Subsequent experiments have indicated that this is more likely due to a survival advantage mediated by IL-7Rα expression in conferring enhanced sensitivity to ambient IL-7 and increased expression of bcl-2, and that CD8+ memory T cells are not selected solely on the basis of IL-7/IL-7Rα interactions, but result from a more complex series of differentiation events, as described subsequently.264,265,266,267,268 Although the IL-7Rαhi effector CD8 T cells preferentially survive following infection compared to IL-7Rαlo cells, there is some death in this population as the memory CD8 T cells form, and therefore this does not represent an absolute determinant of memory T-cell potential.142,249,269 Moreover, not all types of immunizations lead to distinguishable populations of IL-7Rαhi and IL-7Rαlo effector cells.
Memory CD4+ T-cell development may be considered more complicated than that of CD8 T cells because of the diversity in the types of effector T cells formed (eg, Th1, Th2, Th17, Tfh, Treg cells) during immune responses.107 It is unclear if all types of CD4 T cells can generate memory and if so, whether the pathways involved are similar or
distinct. Like CD8 T cells, IL-7Rα is also critical for formation of memory CD4 T-cell populations, and it is similarly repressed in effector CD4 T cells and then reexpressed on memory CD4 T cells during infection.270,271,272 It remains to be determined if IL-7Rαhi CD4+ effector T cells have greater memory cell potential following infection. It is likely that other IL-7Rα-dependent cytokines (such as TSLP) are involved as blockade of IL-7 or STAT5 had little effect on short-term effector CD4 T-cell survival.273,274 Other factors known to regulate activated CD4 T-cell survival long-term are Akt and CD44, one of the most widely used markers for memory T cells.275,276
distinct. Like CD8 T cells, IL-7Rα is also critical for formation of memory CD4 T-cell populations, and it is similarly repressed in effector CD4 T cells and then reexpressed on memory CD4 T cells during infection.270,271,272 It remains to be determined if IL-7Rαhi CD4+ effector T cells have greater memory cell potential following infection. It is likely that other IL-7Rα-dependent cytokines (such as TSLP) are involved as blockade of IL-7 or STAT5 had little effect on short-term effector CD4 T-cell survival.273,274 Other factors known to regulate activated CD4 T-cell survival long-term are Akt and CD44, one of the most widely used markers for memory T cells.275,276
How are heterogeneous populations of effector and memory CD8+ T cells established? It is clear that the duration of infection, level of inflammatory signals, time point at which naïve cells are recruited into the response, and tissue localization can impact both the type of effector T cell generated as well as the memory potential of the activated T cell.142,262,277,278,279,280,281,282 The vast phenotypic and functional heterogeneity observed in antigen-specific CD8+ T-cell populations (as described previously) indicates that this differentiation process is not based on a simple binary cell fate decision, for instance to become a terminal effector cell versus becoming a memory precursor cell. Rather, a more appropriate model for T-cell differentiation may be one in which activated T cells can occupy not just two differentiation states, but a whole spectrum of differentiation states, and these cells can transition from one state to another according to changes in signal input. Indeed, recent in vivo lineage-tracing studies using transgenic mice that express reporter genes indicate that different types of effector CD4+ T cells are likely not fixed lineages as previously thought, but contain cells with greater plasticity that can acquire certain features and functions of other types of CD4+ effector T cells.106,283 For instance, IL-4-producing Th2 cells and IL-17-producing Th17 cells can acquire IFNγ production when restimulated under Th1-conditions, respectively,284,285 and FoxP3+ Tregs can acquire IFNγ and IL-17 expression.286,287,288
Transcriptional Control of Effector and Memory Cell Fates
Although memory T cells share many features with shortlived effectors, their capacity for self-renewal and recall response suggests that they are guided by a distinct program of gene expression. Moreover, the distinction between MPCs and other effector cells that are less fit to populate the memory T-cell pool indicate that variations in effector T-cell differentiation govern these cell fate choices early during the immune response. Investigations into the molecular events that guide the generation memory T cells and their precursors are at a relatively early stage in comparison to the phenotypic studies through which these phenomena have been defined. Nonetheless, a framework for understanding the genetic program of memory is beginning to emerge through the identification of genes and regulatory elements that control key functional parameters of T-cell memory.30,289 Initial studies focused on performing genomescale gene expression analysis in T cells using gene array hybridization techniques, first to resting versus activated T cells, and subsequently to T cells undergoing homeostatic proliferation.31,138,290,291 These studies revealed that the gene expression in T cells is a dynamic process that undergoes substantial alterations in response to TCR-mediated and environmental stimuli during the transition from resting to activated status. The phenotypic features of this process (eg, proliferation, effector differentiation, survival) are matched in the global patterns of genes expressed (eg, cyclins, effector molecules, antiapoptotic factors, etc.).
A key study on the transcriptional profile of memory T cells comes from Kaech et al., who performed genomewide analyses of gene expression on naïve, effector, and memory transgenic CD8+ T cells activated in vivo by LCMV.30 Among the findings emerging from a comparative analysis was that each phase of development was characterized by specific patterns of gene expression directly related to the development or function of the cells at that stage of differentiation, with roughly 30% of the genes expressed by effector cells were also maintained in the memory cells, supporting the direct lineage relationship between these cell types.30 In some cases, the expression level of the genes was set at the peak of the effector response (d8), but in others the level of expression decreased as the gradual conversion to memory occurred, perhaps reflecting a global reduction in transcriptional activity in these cells. Whereas the distinction of TEM and TCM subsets have traditionally been defined by phenotypic markers, Willinger et al. used transcriptional profiling to analyze the molecular signature of these subsets in human CD8+ T cells and found that the TCM subset had a pattern of gene expression that was intermediate between naïve and TEM and effector/TEM phenotype cells.292 It has long been appreciated that memory T cells share several notable features related to self-renewal with long-term hematopoietic stem cells. Consistent with this, Luckey et al. have found that memory T cells appear to share many elements of their transcriptional program with long-term hematopoietic stem cells, with notable increases in the expression of prosurvival and antiapoptosis genes.293
In parallel with the genomic-scale studies of gene expression at the population level in T cells, efforts have been made to define the genetic program of memory in single cells or with respect to specific transcription factors. The recent study of Peixoto et al. is notable in its study of gene expression patterns at the level of single cells at various stages during an immune response.294 This study revealed surprisingly heterogeneous and dynamic patterns of gene expression from the earliest point in the response, with both proinflammatory and effector functionality revealed in discrete cells, and was followed by a more uniform pattern in secondary responders. It is unclear if the reduction in heterogeneity at later time points reflects a selection process or progressive development of the entire memory populations. A number of studies have analyzed the contribution of specific transcription factors to the enhanced longevity and function of memory T cells. These include T-bet and eomesodermin, the transcription factors that coordinately regulate a number of genes related to lineage commitment and effector differentiation in both CD4+ and CD8+ T cells.108,109,295,296,297 T-bet-/-
eomesodermin+/- mice have a near-total loss of effector and memory T cells through a mechanism that is believed to involve their role in regulating the expression of CD122 and thereby responsiveness to IL-15.298 The roles of T-bet and eomesodermin in determining whether effector cells are selected for memory development may involve their reciprocal regulation in response to inflammatory signals via IL-12. In response to pathogen-induced IL-12, eomesoderminis repressed and T-bet levels increase which can lead to inhibition of IL-7Rα and MPCs, but increased clonal expansion and formation of terminal effector cells.50,299,300 When IL-12 signals are reduced, eomesodermin levels rise while T-bet levels decrease, favoring the development of long-lived MPCs.142,299 Another pair of transcription factors that are relevant for memory formation is inhibitor of deoxyribonucleic acid binding 2 (Id2), and Id3, antagonists of E protein transcription factors that are upregulated in CD8+ T cells during infection and appear to have somewhat reciprocal expression patterns and temporal modes of actions.301,302 Id2 plays in important early during clonal expansion in sustaining viability of proliferating CD8+ T cells, and it is expressed to a higher level in terminal effector CD8 T cells, whereas Id3 is expressed to a higher extent in memory precursor cells and is required for their survival following infection during the effector to memory transition.301,302,303 Id2- and Id3-deficient CD8+ T cells have altered patterns of expression in genes that influence cell survival, but they appear to operate on distinct sets of genes.301,302,303
eomesodermin+/- mice have a near-total loss of effector and memory T cells through a mechanism that is believed to involve their role in regulating the expression of CD122 and thereby responsiveness to IL-15.298 The roles of T-bet and eomesodermin in determining whether effector cells are selected for memory development may involve their reciprocal regulation in response to inflammatory signals via IL-12. In response to pathogen-induced IL-12, eomesoderminis repressed and T-bet levels increase which can lead to inhibition of IL-7Rα and MPCs, but increased clonal expansion and formation of terminal effector cells.50,299,300 When IL-12 signals are reduced, eomesodermin levels rise while T-bet levels decrease, favoring the development of long-lived MPCs.142,299 Another pair of transcription factors that are relevant for memory formation is inhibitor of deoxyribonucleic acid binding 2 (Id2), and Id3, antagonists of E protein transcription factors that are upregulated in CD8+ T cells during infection and appear to have somewhat reciprocal expression patterns and temporal modes of actions.301,302 Id2 plays in important early during clonal expansion in sustaining viability of proliferating CD8+ T cells, and it is expressed to a higher level in terminal effector CD8 T cells, whereas Id3 is expressed to a higher extent in memory precursor cells and is required for their survival following infection during the effector to memory transition.301,302,303 Id2- and Id3-deficient CD8+ T cells have altered patterns of expression in genes that influence cell survival, but they appear to operate on distinct sets of genes.301,302,303
In addition to the transcription factors described previously, several transcriptional repressors have emerged as regulators of T-cell memory. Bcl-6, first described as a transcriptional repressor with pleiotropic functions in B-cell differentiation,304 appears to play a role the generation of both CD4+ and CD8+ T-cell memory, as the survival of Bcl-6-deficient CD4+ effector T cells was greatly diminished compared to wild-type controls after priming, and Bcl-6-deficient mice were found to lack TCM memory phenotype CD8+ T cells following immunization.305,306,307 The Bcl-6 homologue Bcl-6b, known to repress IL-2-induced B-cell differentiation, is expressed by a small subset of CD8+ T cells and regulates their capacity for secondary expansion in response to antigenic challenge.308 Another transcriptional repressor first described in B cells that influences T cell homeostasis is Blimp-1, which is required for terminal differentiation of B cells through coordinate regulation of hundreds of genes.309,310 Mice lacking Blimp-1 suffer from a variety of immune-mediated pathologies including colitis and multiorgan inflammatory diseases that are characterized by accumulation of effector and memory T cells.311,312 Moreover, Blimp-1 represses Id3 expression and promotes the formation of highly functional, yet terminal, IL-7Rαlo CTLs during viral infection.301,313,314 In the absence of Blimp-1, many more IL-7Rαhi memory CD8 T cells and their precursors form; however, these cells are less efficient in migrating out of the lymphoid organs and defending against respiratory infection. Therefore, the Blimp-1:Bcl-6 axis is an important regulator of effector and memory CD8 T-cell fates. Interestingly, the balance between Blimp-1 and Bcl-6 also regulates TH1:follicular helper T cell fates during viral infection.128
Phase III. Maintenance and Function of T-Cell Memory
The small fraction of effector T cells that survives contraction will initiate the memory pool that confers, in many cases, lifelong protection to the individual. Despite their extensive culling from peak numbers, antigen-specific cells in the postcontraction population are found at a far higher precursor frequency than in the naïve repertoire, a fact that undoubtedly impacts the increased speed and intensity that is characteristic of secondary responses. Although they possess some features associated with memory, having arisen in response to an antigenic challenge and being able to persist after the majority of their fellow effectors have been eliminated, the postcontraction T cells nonetheless comprise a heterogeneous population whose maintenance in the memory pool is a dynamic process involving phenotypic changes that will determine their function, distribution, and protective capacity over time.36,42,315 A central theme governing this process involves adaptation to the homeostatic control mechanisms that regulate the equilibrium of naïve versus memory T cells. These pathways appear to be distinct and under independent control, as the homeostatic mechanisms that regulate the naïve and memory T-cell pools differ in their requirement for extrinsic factors such as TCR stimulation by self-MHC molecules (presumably occupied with peptide derived from self- or environmental antigens) and signals provided by the γc cytokines IL-7 and IL-15.
T cells sense each other and maintain homeostasis by competing with one another for survival signals, likely in the confines of cellular niches within tissues. When the T-cell compartment is “full,” as normally found in lymphoreplete states, naïve T cells are fairly quiescent and memory T cells divide periodically, approximately once every 2 to 4 weeks. This slow turnover of memory T cells is referred to as “basal proliferation” or “homeostatic turnover,” and is thought to be important for maintaining the pool of memory T cells.259 In contrast, in lymphopenic states, such as following irradiation or transfer into immunocompromised mice lacking T cells (ie, recombination activating gene-deficient mice) both naïve and memory T cells will divide more rapidly to fill the available “space.” This “homeostatic expansion” more commonly referred to as “homeostatic proliferation” is polyclonal and occurs independent of foreign antigen recognition. For the purposes of this section, we shall refer to the slow turnover of T cells in a normal immunocompetent host as “basal homeostatic proliferation” and the faster, more extensive division of T cells within lymphopenic hosts as “homeostatic expansion.”
The Role of Major Histocompatibility Complex in Memory T-Cell Homeostasis
As discussed previously in this chapter, adoptive transfer studies have shown that the long-term survival of naïve T cells in lymphopenic hosts requires contact with self-MHC.62,63,64,65,66,67 Whereas initial studies in the 1990s suggested that both types of memory cells were maintained by occasional TCR contact with undetectable amounts of sequestered foreign or cross-reactive environmental antigens,
more recent studies have shown that antigen-specific CD8+ memory T cells can survive and remain functionally competent in the absence of either antigen, TCR signaling adaptors, or MHC molecules, arguing against any obligatory role for TCR signaling in their maintenance.32,67,316,317,318,319,320,321 However, the same may not be true during cases of chronic antigen exposure, such as during chronic infections. One study has demonstrated that virus-specific CD8 T cells isolated from mice with a chronic LMCV infection require antigen for their persistence,145 and a similar situation may be evidenced after control of viremia by highly active antiretroviral therapy in patients with HIV.322,323 Moreover, in another study, inducible ablation of the TCR on mature MP T cells showed that MP CD8+ T cells declined slowly over time, possibly indicating differences between antigen-derived versus MP T cells. The situation for CD4+ memory T cells is less clear, because while the antigen-specific CD4+ T cells do not strictly depend on MHC for their maintenance, the rate of both basal homeostasis and homeostatic expansion of MP CD4+ T cells does appear to depend on the TCR-MHC interactions, and declines significantly when these signals are extinguished, leaving the cells in one study critically dependent on IL-7 or less responsive to antigen reencounter in vitro and in vivo.62,324,325,326,327,328,329 Thus, it is likely that the homeostasis of memory CD4 and CD8 T cells depends on some common, but also distinct, signals.
more recent studies have shown that antigen-specific CD8+ memory T cells can survive and remain functionally competent in the absence of either antigen, TCR signaling adaptors, or MHC molecules, arguing against any obligatory role for TCR signaling in their maintenance.32,67,316,317,318,319,320,321 However, the same may not be true during cases of chronic antigen exposure, such as during chronic infections. One study has demonstrated that virus-specific CD8 T cells isolated from mice with a chronic LMCV infection require antigen for their persistence,145 and a similar situation may be evidenced after control of viremia by highly active antiretroviral therapy in patients with HIV.322,323 Moreover, in another study, inducible ablation of the TCR on mature MP T cells showed that MP CD8+ T cells declined slowly over time, possibly indicating differences between antigen-derived versus MP T cells. The situation for CD4+ memory T cells is less clear, because while the antigen-specific CD4+ T cells do not strictly depend on MHC for their maintenance, the rate of both basal homeostasis and homeostatic expansion of MP CD4+ T cells does appear to depend on the TCR-MHC interactions, and declines significantly when these signals are extinguished, leaving the cells in one study critically dependent on IL-7 or less responsive to antigen reencounter in vitro and in vivo.62,324,325,326,327,328,329 Thus, it is likely that the homeostasis of memory CD4 and CD8 T cells depends on some common, but also distinct, signals.
The Role of Common γ-Chain Cytokines in Memory T-Cell Homeostasis: Interleukin-7 and Interleukin-15
These findings of MHC-independent survival of memory T cells have brought cytokines into focus as the main extrinsic factors responsible for their maintenance, with particular emphasis on the common γc family members IL-7 and IL-15. One of the important initial observations linking cytokines with memory T-cell survival came from studies in which agents capable of stimulating IFN production by cells of the innate immune system, such as lipopolysaccharide and Poly I:C caused a transient burst of TCR-independent proliferation from the major CD122hi CD44hi subset of antigen-experienced CD8+ T cells through the IFNαβ-induced production of IL-15.330,331,332,333,334,335 Importantly, the IFN-induced proliferative burst of CD122hi CD44hi CD8+ T cells was not observed in IL-15-/- mice, and the in vivo administration of exogenous IL-15 was found to mimic the effect of IFNs in stimulating the bystander proliferation of the MP CD8+ T-cell subset.331,336,337
The receptor for IL-15 is composed of three subunits: a unique α chain, a β chain shared with the IL-2 receptor and γc, the latter sheared with the receptors for with IL-2, IL-4, IL-7, IL9, and IL-21.338,339,340 Although a soluble cytokine under in vivo conditions, IL-15 is presented in trans via cell-associated complexes bound to IL-15Rα that are preassembled in the cytoplasm of the (non-T) cells that make both molecules.341,342,343 Paradoxically, CD8+ T cells also express IL15Rα, and although this is not essential for them to respond to IL-15 (which requires expression of only the β and γ chains), it may enable them to activate other cell types such as T cells or APC via transpresentation.341,342,344,345 The expression of CD122 is selectively elevated on the MP (CD44hi) subset of CD8+ T cells that undergo bystander proliferation in response to IL-15, and both IL-15-/- and IL-15Rα-/- mice contain significantly reduced numbers of CD44hi CD122hi MP CD8+ T cells.331,346,347,348 The effect of IL-15 on MP CD8+ T cells may reflect its role in cell survival rather than development, as these fail to proliferate and disappear rapidly upon transfer to IL-15-/- recipients.331 Consistent with this view, IL-15 transgenic mice were found to contain elevated numbers of MP CD8+ T cells.337,349 Taken together, these findings have led to the current view that the generation, survival, and basal homeostatic proliferation of MP CD8+ T cells is mediated through contact with ambient IL-15. At steady state, DCs and macrophages are the most important cell types that need to express IL-15:IL-15Rα complexes to sustain memory CD8 T cells in lymphoid organs, and intestinal epithelial cells are critical in the gut.217 However, this does not exclude the production of IL-15 by a variety of other cell types including skeletal muscle, kidney, placenta, and hematopoietic stromal cells under conditions of innate immune activation via the IFN pathway.334,350,351
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


