years in westernized countries.6,7,8 In such countries, 30% of the population manifest some form of atopic disease at some time in their lives. Interestingly, the low baseline level of atopic diseases in developing countries has not changed over the same time period, suggesting that factors associated with the westernized lifestyle predispose to atopic disease. The widespread prevalence and morbidity of atopic diseases imposes a heavy burden on society.
TABLE 45.1 Definitions of Key Terms | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||
TABLE 45.2 Major Features of Allergic Immune Responses | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||
may persist for days in the absence of therapy. The pathophysiologic consequences of chronic reactions are associated with the migration of eosinophils and lymphocytes from the blood into affected tissues.
relatively high levels of proteins (approximately 50 µg) in venom are injected subcutaneously during a sting. The venom-associated allergens of several vespids (yellow jacket, wasp, fire-ants, and white faced hornet) are cross-reactive and include antigen 5, phospholipase, and hyaluronidase. The honeybee venom contains distinct allergens, including two major ones, phospholipase A2 and hyaluronidase, and a less important one, melittin. Many of these allergens have proteolytic activity.
a generalized increase in T-cell activation in all disorders of the atopic triad, with increased expression of the IL-2 receptor, class II histocompatibility antigens (human leukocyte antigen [HLA]-DR), and very late activation antigen-1.28,30 These activated T cells have the capacity to rapidly expand in response to specific stimuli, and through the release of a variety of cytokines they recruit and activate other immune cells (B cells, CD8 T cells, macrophages, mast cells, neutrophils, eosinophils, basophils) thereby initiating a complex series of events resulting in the symptoms of allergic diseases. Following initial activation, CD4+ T cells remain in lymphoid tissues as memory cells. These memory cells retain the ability to respond to specific antigens upon reexposure throughout the lifetime of the individual.
and atopic dermatitis.58 Collectively, the Th2 cytokines orchestrate the elicitation of the allergic response via their ability to regulate IgE production and recruitment and activation of various effector cells (eg, mast cells, eosinophils). In particular, IL-4, through its critical role in Th2 differentiation, has been shown to be essential in the initiation of allergic airway responses.59,60 IL-5 clearly plays a role in eosinophil development, recruitment, and activation at the site of Th2-inflammatory responses,61 whereas IL-9 appears to be an important regulator of mast cell activation.62 IL-13 has been shown to have a singular role in the effector phase of the allergic response.63,64 Specifically, it is sufficient to induce many of the manifestations of allergic disease including airway inflammation, AHR, and mucus cell hypersecretion in allergic airway diseases.
severe asthma.81,82 Interestingly, exposure to many known triggers of asthma exacerbations, including organic dust, inhaled particulate matter, and ozone, have been shown to trigger release of Th17 cytokines.83,84,85
of antigen stimulation. These antigen-specific Tregs secrete anti-inflammatory cytokines such as IL-10 and TGF-β and regulate immune responses and inflammatory pathologies. Antigen-induced Tregs that secrete IL-10 are often referred to as IL-10-Treg cells, or Tr1 cells, whereas those that secrete TGF-β have been referred to as Th3 cells.
complex genetic nature of atopic disorders, multiple studies have reported evidence of linkage of atopic related traits with five primary chromosomal regions including 1) the Th2 cytokine gene cluster on 5q31-q33 including the IL-4, IL-13, CD14, SPINK5, LTC4S, and CYFIP2 genes; 2) the HLA region on 6p21 including both classical (HLA-DRB1, HLADQB1, HLA-G) and nonclassical HLA genes (TNF, LTA); 3) the region that contains the high-affinity IgE receptor gene (FcεRIb) on 11q13; 4) a large region on chromosome 12q14 that spans several candidate genes (Stat6, IFNγ, stem cell factor, nitric oxide synthase 1); and 5) a region of 16q containing the IL4RA.127 Disease-specific genes have also been identified through candidate gene approaches and genome-wide association studies: asthma (IL33, IL18R1, IL2RB, SMAD3, ORMDL3, HLA-DAQ),128 AD (the epithelial barrier gene, filaggrin),129 and eosinophilic eosphagitis (thymic stromal lymphopoietin [TSLP]).130
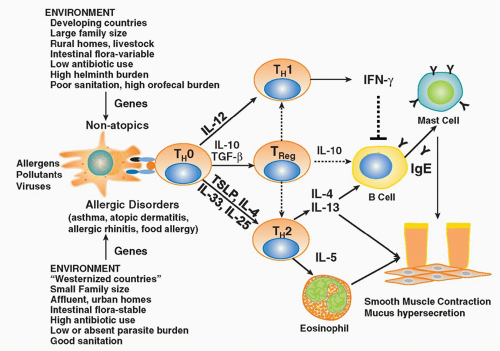 FIG. 45.1. Allergic Diseases are Associated with Aberrant Adaptive Immune Responses to Innocuous Environmental Antigens. The development of allergic responses to harmless antigens is influenced both by environmental factors and the genetic background of the individual. Under conditions in which microbial exposure is minimal such as in developed countries, T cells differentiate along a Th2 pattern with elaboration of interleukin (IL)-4, IL-13, IL-5, and IL-9. In contrast, microbial exposure in early life protects against the development of harmful Th2 immune responses via the induction of regulatory T cells. |
of allergen-specific T-cell responses may occur before birth.134,135 It has been demonstrated that the responding cord blood T cells were indeed of fetal origin, as they did not respond to common vaccine antigens such as tetanus toxoid, which the majority of adults in the study population expressed active immunity against.134 These findings suggest that transfer of substances from mother to child (eg, allergens, antibodies, toxins, hormones, or other immune mediators via the placenta) may occur during gestation, which might prime or sensitize the developing infant to environmental antigens. In addition, factors present in the in utero environment may serve to influence the nature of the immune response of the fetus to allergens that cross the placenta. Indeed, mediators such as PGE2 and high levels of progesterone, which has been shown to alter the Th1/Th2 balance by either suppressing Th1 cytokine production or inducing a Th2 pattern, are present during pregnancy. Th2 skewing is thought to take place to suppress IFNγ production that is toxic to the fetus. Accordingly, exogenous antigens that leak across the placenta are likely to be presented to the fetal immune system within a milieu conducive to selection for Th2 immunity. This contention is supported by studies showing that cord blood cells from both allergic and noallergic infants both produce high levels of Th2 cytokines and low IFNγ levels.135,136 Although immune responses in neonates are skewed toward a Th2 phenotype, studies of human infants indicate that this Th2 skew gradually diminishes during the first 2 years of life in nonallergic individuals.137 In allergic infants, the reverse occurs, with the strength of neonatal Th2 responses increasing over a similar period. This has been supported by Holt and colleagues,138 who showed that although the levels of IgE vary considerably during early childhood, atopic subjects eventually lock into a stable pattern of increasing antibody production and Th2-polarized cellular immunity that is associated with stable expression of the IL-4 receptor in allergen-specific Th2 memory cells, which is absent during infancy. The persistence of this neonatal bias and the failure to produce Th1 or Treg-type responses may be an important feature of the atopic disease state.
among children of mothers with asthma but with modest risk for asthma among children of mothers without asthma. The mechanism through which the “maternal asthma-fetal HLA-G genotype” interaction influences subsequent risk for asthma in the child is still unknown, but modulation of expression via miRNA targeting could be involved. Because asthma in the mother is such a strong predictor of asthma in her children, it is likely that other, as yet undiscovered, genes also play a role in modulating this effect.
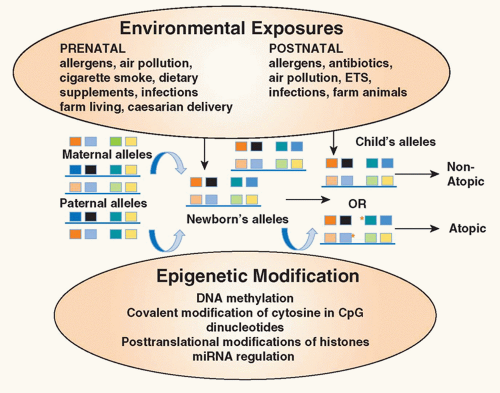 FIG. 45.2. Environmental Epigenetics and Atopy. Exposure of the mother to environmental factors (environmental tobacco smoke, air pollution, allergens, antibiotics, dietary supplements) during pregnancy may induce epigenetic changes (deoxyribonucleic acid methylation, covalent modification of cytosine in cytosin-phosphatidyl-guanosin dinucleotides, posttranslational modifications of histones, micro ribonucleic acid regulation) in gene expression in the offspring providing an explanation for the heretofore unexplained maternal influence on the inheritance of atopic diseases. In addition, exposure of the infant in early childhood to environmental factors may also alter expression of genes important in allergic inflammation. Collectively, these influences contribute to the risk of the child developing atopic diseases. Red asterisks refer to sites of epigenetic changes. Adapted from Miller and Ho.164 |
3) activation of various danger sensing pathways (extracellular nucleotides, uric acid).
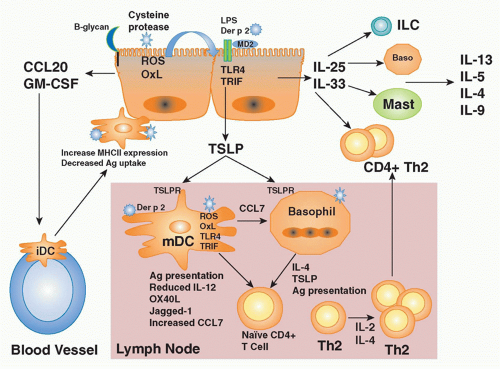 FIG. 45.3. Schematic of Events Leading to the Initiation of Th2-Mediated Immune Responses. Upon encounter with allergen, epithelial cells of the respiratory tract, the intestinal tract, and the skin release mediators that recruit (CCL20), activate (granulocyte macrophage-colony stimulating factor [GM-CSF]), and/or drive Th2 cell differentiation (thymic stromal lymphopoietin [TSLP], interleukin [IL]-25, IL-33). IL-25 and IL-33 recruit and activate a variety of innate immune cells such as basophils, mast cells, and innate lymphoid cells, which can produce the protypical type 2 cytokines IL-4, IL-5, and IL-13 driving the allergic phenotype. The elevations in IL-25 and IL-33 can perpetuate the allergic response by further type 2 cytokine production from basophils, eosinphils, or by initiating conventional Th2-cell expansion through activating dendritic cells (DCs) during encounters with allergens. In parallel, immature myeloid DCs are recruited to mucosal surfaces following the release of chemokines (ie, CCL20) and cytokines (GM-CSF) by epithelial cells in response to inflammatory stimuli such as allergens, pollutants, irritants, and/or viral infections. Once in the tissues, DCs mature and express costimulatory molecules (ie, CD86, OX40L, Jagged-1) under the influence of cytokines (TSLP, IL-33, IL-25) derived from surrounding tissues through stimulation of pattern recognition receptors (likely TLR4, -2; C-type lectins) expressed on their surface. In atopic individuals, it has been postulated that myeloid DCs appear to be the predominant population of DCs taking up antigen and driving the differentiation of naïve T cells toward a Th2 profile. They then migrate to the draining lymph node to stimulate naïve T-cell differentiation and proliferation. Proteasecontaining allergens induce the production of TSLP by epithelial cells through a series of steps including generation of reactive oxygen species, production of oxidized lipids, which in turn stimulate TLR4-TRIF signaling pathways leading to TSLP expression. These same pathways induce the production of the chemokine CCL7 by DCs, which recruits basophils to the lymph node. In the lymph node, they can also directly stimulate basophils to produce TSLP and IL-4. Basophils and DCs drive Th2 cell differentiation under the influence of various cytokines such as TSLP, IL-4, and IL-25. These Th2 cells proliferate in response to IL-2 and IL-4 in an autocrine fashion. Activated Th2 cells in turn migrate back to the site of antigen stimulation under the influence of Th2 selective chemokines (CCL17, CCL12). Once in the tissues, Th2 cells secrete a profile of Th2 cytokines (IL-4, IL-5, IL-13, IL-9) that initiate a cascade of downstream pathways that collectively lead to the development of the allergic phenotype. Not shown on the figure is the fact that through their effects on the airway epithelium, Th2 cytokines may further amplify Th2 immune responses through the induction of Th2-selective chemokines (CCL17, CCL12) and/or IL-25 and IL-33 initiating a positive feedback loop. baso, basophil; iDC, immature dendritic cell; ILC, innate lymphoid cell; mDC, myeloid dendritic cell; ROS, reactive oxygen species. |
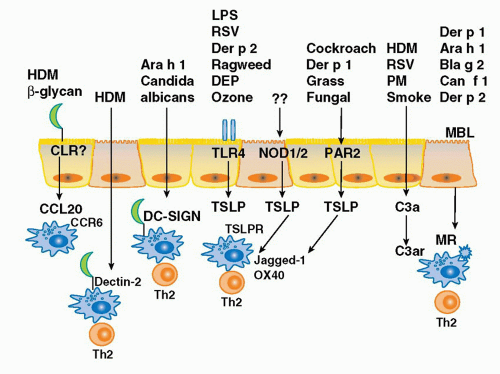 FIG. 45.4. Innate Immune Pathways Activated by Allergens. Allergenic peptides are recognized by several traditional pattern receptors including toll-like receptors, C-type lectin receptors, and nucleotide oligomerization domain receptors. In addition, other classes of allergens initiate Th2-mediated immune responses through the activation of protease-activated receptors and the cleavage of complement pathways. Activation of these innate immune pathways on mucosal epithelial cells leads to the production of chemokines which recruit immature demdritic cells (DCs), the production of Th2-promoting cytokines such as thymic stromal lymphopoietin, the enhancement of DC internalization of allergens, and the upregulation of expression of costimulatory molecules associated with Th2 differentiation such as OX40L and jagged-1. DEP, diesel exhaust particles; HDM, house dust mite; MBL, mannan-binding lectin; MR, mannose receptor; PM, particulate matter. Der p 1 and Der p 2 are house dust mite-derived allergens; Ara h 1 is the major peanut allergen; Blag 2 is the major cockroach allergen; Can f 1 is the major canine allergen. |
of the TLR4 signaling complex, Trompette and colleagues25 asked the question whether Der p 2 and MD-2 exhibited functional homology as well. Indeed, they reported that Der p 2 facilitates TLR4 signaling through direct interactions with the TLR4 complex, reconstituting LPS-promoted TLR4 signaling in the absence of MD-2. They also demonstrated that Der p 2 could facilitate LPS signaling in primary APCs, with or without MD-2 being present. Finally, they reported that in vivo delivery of Der p 2 drives the development of experimental allergic asthma in a TLR4-dependent manner, retaining this property in mice with a genetic deletion of MD-2. These data suggest the possibility that Der p 2-mediated activation of TLR4 signaling in the airway epithelium under conditions of low bacterial product exposure—those associated with increasing rates of aeroallergy in the urban, developed world— may shift the LPS/TLR4 response curve from the tolerizing into the Th2-inducing range. Der p 2 exposure may serve to facilitate TLR4 signaling by airway epithelial cells as they have been reported to express TLR4, but little or no MD-2, in the basal state.185
of defense against a large number of microbial pathogens including bacteria, fungi, protozoa, and viruses. Indeed, it has recently been shown that the major cat allergen, Fel d 1, is a novel ligand of the cysteine-rich domain of the mannose receptor and the development of allergic inflammation induced by Fel d 1 was attenuated in mannose receptor-deficient mice.192 Likewise the mannose receptor has been shown to mediate the internalization of a diverse range of allergens (Der p 1, Der p 2, Can f 1, Bla g 2, Ara h 1) into monocyte-derived DCs through their carbohydrate moieties.193 Alterations in these pathways may lead to enhanced recognition of allergens as sequence variations in the MRC1 gene have recently been associated with the development of asthma in two independent and ethnically diverse populations (Japanese and African American).194
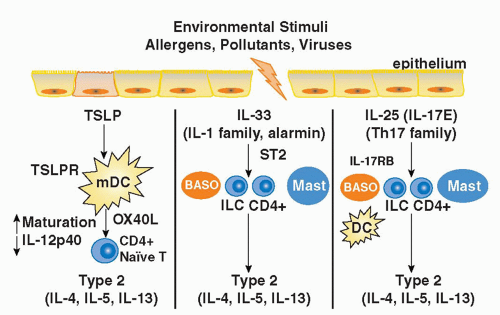 FIG. 45.5. Members of Three Distinct Cytokine Families Promote Th2 Differentiation. Thymic stromal lymphopoietin (TSLP), an interleukin (IL)-7-like cytokine released from mucosal epithelial cells, binds its receptor TSLPR on dendritic cells and induces the expression of OX40L and the suppression of IL-12 production, thus driving the differentiation of naïve T cells toward a Th2 phenotype. IL-33 is an IL-1 family member that binds its unique receptor ST2 on a variety of innate immune cells including basophils, innate lymphoid cells, conventional CD4+ T cells, and mast cells driving the production of type 2 cytokines. IL-25, a member of the IL-17 cytokine family, binds its receptor IL-17RB on a variety of innate immune cells including basophils, innate lymphoid cells, mast cells, and conventional CD4+ T cells driving their production of type 2 cytokines. IL-25 and IL-33 can also stimulate type 2 cytokine production indirectly by activating dendritic cells to promote Th2 differentiation. |
IL-1 receptor accessory protein (IL-1RAcP),223 both of which are widely expressed, particularly by innate immune cells (mast cells, basophils, DCs), Th2 cells, and innate lymphoid populations described previously.217,223 IL-33 is thought to function as an “alarmin” released from stromal cells (epithelial cells, endothelial cells, and airway smooth muscle) following cellular injury to alert the immune system to tissue damage or stress. A role for IL-33 in type 2-cytokine dependent immune responses in vivo is supported by the finding that administration of IL-33 to naive mice induces eosinophilia, increased serum IgE levels, IL-5 and IL-13 production, and mucus cell changes in the respiratory and gastrointestinal tracts.224 Moreover, blocking IL-33 with an ST2 fusion protein decreases eosinophilic airway inflammation, concomitant with a decrease in IL-4 and IL-5 expression.225 Notably, the levels of soluble ST2 protein and IL-33 mRNA protein are increased in sera and tissues from patients with asthma.226,227 Genome-wide association studies have identified polymorphisms in the genes encoding Il1rl1 and Il33 in patients with asthma, hypereosinophilic diseases, and allergic rhinitis, suggesting an association with atopic disorders in general.228,229,230
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


