Immunoglobulins: Structure and Function
Harry W. Schroeder Jr
David Wald
Neil S. Greenspan
INTRODUCTION
Immunoglobulins (Igs) are marked by a duality of structure and function.1 In common with other members of the Ig superfamily,2 they provide the immune system with a conserved set of effector molecules. These effectors can activate and fix complement and they can bind to Fc receptors on the surfaces of granulocytes, monocytes, platelets, and other components of the immune response. Both activation of complement and binding to Fc receptors can contribute to the induction or maintenance of inflammation. They also provide the immune system with a polyclonal set of diverse ligand binding sites, which allow Igs, as a population, to recognize an almost unlimited array of self- and non-self-antigens, which may range from compounds as fundamental to life as deoxyribonucleic acid to manmade molecules that could not have played a role in the evolution of the immune system.
Differential splicing allows individual Ig molecules to serve as either membrane-bound receptors for the B cell that allow antigen-specific activation or as soluble effectors, which act at a distance. In vivo, proper effector function requires more than just antigen-specific binding; it requires successful neutralization of the offending antigen while avoiding potentially pathogenic self-reactivity.
The receptor and effector functions of each individual Ig can be localized to a separate region or domain of the molecule. Each variable (V) or constant (C) domain consists of approximately 110 to 130 amino acids, averaging 12,000 to 13,000 kD. A typical light (L) chain will thus mass approximately 25 kD, and a three C domain Cγ H chain with its hinge will mass approximately 55 kD.
Immunoglobulins are Heterodimers
Igs are heterodimeric proteins, consisting of two H and two L chains (Figs. 5.1 and 5.2). The eponymous Ig domain serves as the basic building block for both chains. Each of the chains contains a single amino-terminal V Ig domain and one, three, or four carboxy-terminal C Ig domains. H chains contain three or four C domains, whereas L chains contain only one. H chains with three C domains tend to include a spacer hinge region between the first (CH1) and second (CH2).3
At the primary sequence level, Igs are marked by the interspersion of regions of impressive sequence variability with regions of equally impressive sequence conservation. The V domains demonstrate the greatest molecular heterogeneity, with some regions including non-germline-encoded variability and others exhibiting extensive germline conservation across 500 million years of evolution.4 The molecular heterogeneity of the V domains permits the creation of binding sites, or paratopes, which can discriminate between antigens that may differ by as little as one atom. Thus it is the V domains that encode the receptor function and define the monovalent specificity of the antibody. The H chain CH1 domain, which is immediately adjacent to the V, associates with the single L chain C domain. Together, the CH1 and CL domains provide a stable platform for the paired set of VH and VL domains, which create the antigen binding site. The distal CH2 and CH3 domains, for those antibodies with a hinge, or the CH3 and CH4 domains, for those with an extra (CH2) domain, typically encode the effector functions of soluble antibody. Each of these Ig CH domains is encoded by a separate exon. Although the sequences of the individual CH domains are constant within the individual (and nearly constant within a species), they can vary greatly across species boundaries. The carboxy-terminal CH domain encodes a secretory tail, which permits the antibody to exit the cell.
Also encoded within the germline sequence of each CH gene are two membrane/cytoplasmic tail domain exons, termed M1 and M2. Alternative splicing removes the secretory sequence typically encoded by the terminal CH3 or CH4 domain and replaces it with the peptides encoded by the M1 and M2 exons, converting a secretory antibody to a
membrane-embedded receptor.5 Between species, the membrane/cytoplasmic tail region is the most highly conserved portion of the CH domains, which befits its role as a link to the intracellular signal transduction pathways that ultimately regulate B-cell function.
membrane-embedded receptor.5 Between species, the membrane/cytoplasmic tail region is the most highly conserved portion of the CH domains, which befits its role as a link to the intracellular signal transduction pathways that ultimately regulate B-cell function.
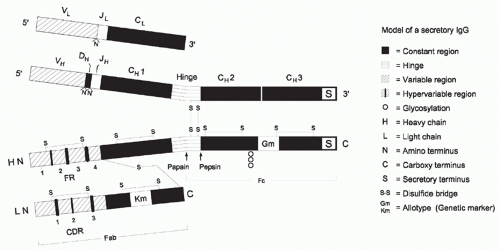 FIG. 5.1. Model of a Secreted Immunoglobulin (Ig)G. The germline exonic derivation of the sequence is shown at the top, and the protein structure is shown at the bottom. The location of the various cysteine residues that help hold both the individual domains and the various Ig subunits together are illustrated. Papain digests IgG molecules above the cysteine residues in the hinge that holds the two H chains together yielding two Fab molecules and an Fc, whereas pepsin digests below releasing an (Fab)′2 fragment and two individual Fcs (which are typically degraded to smaller peptide fragments). The location of some allotypic variants is illustrated. CH2 domains can be variably glycosylated, which can also affect Ig structure and effector function. |
Paratopes and Epitopes
The immunoglobulin-antigen interaction takes place between the paratope, the site on the Ig at which the antigen binds, and the epitope, which is the site on the antigen that is bound. It is important to appreciate that antibodies do not recognize antigens; they recognize epitopes borne on antigens.6 This makes it possible for Igs to discriminate between two closely related antigens, each of which can be viewed as a collection of epitopes. It also is one scenario that permits the same antibody to bind divergent antigens that share equivalent epitopes, a phenomenon referred to as cross-reactivity.
It has been estimated that triggering of effector functions in solution typically requires aggregation of three or more effector domains, and thus tends to involve the binding of three or more epitopes.6 For antigens encoding repeating epitope structures, such as polysaccharides or antigen aggregates, binding of a single polymeric Ig molecule carrying multiple effector domains, such as pentameric IgM, can be sufficient to induce effector function. For antigens encoding diverse epitopes, which is more typical of monodisperse single-domain molecules in solution, triggering of inflammatory effector functions may require the binding of a diverse set of Ig molecules, all binding the same antigen, but at different epitopes.7
Membrane and Secretory Immunoglobulin
Alternative splicing allows Igs to serve either as soluble antibodies or as membrane-bound antigen receptors. In their role as antibodies, Igs are released into the circulation from where they may traffic into the tissues and across mucosal surfaces. In their role as the B-cell antigen receptor, they are anchored to the membrane by means of their M1:M2 transmembrane domain. Soluble antibodies can also be pressed into service as heterologous cell surface antigen receptors by means of their attachment to membrane-bound Fc receptors.8 This permits the power of antibody recognition to be extended to nonlymphoid cells, such as Fc-expressing granulocytes, macrophages, and mast cells. The major difference between these two forms of cell surface receptors is that Igs as B-cell antigen receptors provide a monoclonal receptor for each B cell, whereas antibodies bound to Fc receptors endow the cell with a polyclonal set of antigen recognition molecules. This gives greater flexibility and increases the power of the effector cells to recognize antigens with multiple non-self-epitopes.
Isotypes and Idiotypes
Igs can also serve as antigens for other Igs. Immunization of heterologous species with monoclonal antibodies (or a restricted set of Igs) has shown that Igs contain both common
and individual antigenic determinants. Epitopes recognized within the V portion of the antibodies used for immunization that identify individual determinants are termed idiotypes (Fig. 5.3), whereas epitopes specific for the constant portion are termed isotypes. Recognition of these isotypes first allowed grouping of Igs into recognized classes. Each class of Ig defines an individual set of C domains that corresponds to a single H chain constant region gene. For example, IgM utilizes µ H chain C domains and IgE utilizes ε C domains.
and individual antigenic determinants. Epitopes recognized within the V portion of the antibodies used for immunization that identify individual determinants are termed idiotypes (Fig. 5.3), whereas epitopes specific for the constant portion are termed isotypes. Recognition of these isotypes first allowed grouping of Igs into recognized classes. Each class of Ig defines an individual set of C domains that corresponds to a single H chain constant region gene. For example, IgM utilizes µ H chain C domains and IgE utilizes ε C domains.
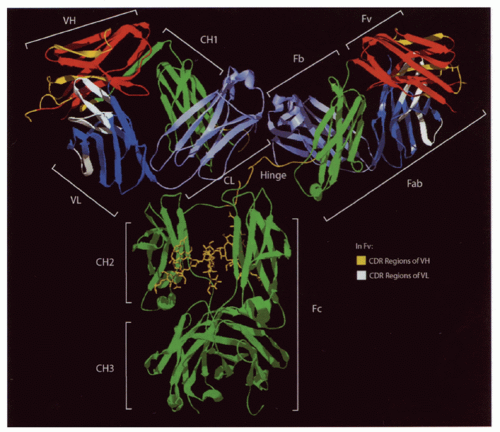 FIG. 5.2. Ribbon Diagram of a Complete Immunoglobulin (Ig)G1 Crystal (1 hzh in the protein data bank [PDB] from Data of Harris et al.202). The major regions of the Ig are illustrated. The heavy-chain constant regions (green) also include the hinge (yellow) between the first two domains. Cγ2 is glycosylated (also seen in yellow). The heavy- and light-chain variable regions (red and dark blue, respectively) are N terminal to the heavy- (green) and light-chain (light blue) constant regions. Complementarity determining region loops in the heavy- and light-chain variable regions (yellow and white) are illustrated as well. |
Some V domain epitopes derive from the germline sequence of V gene exons. These shared epitopes, commonly referred to as public idiotopes or cross-reactive idiotypes (see Fig. 5.3), are, from a genetic perspective, isotypic because they can be found on many Igs of different antigen-binding specificities that derive from the same germline V region. Examples include the cross-reactive idiotypes found on monoclonal IgM rheumatoid factors derived from individuals with mixed cryoglobulinemia, each of which can be linked to the use of individual V gene segments.9
Classes and Allotypes
Each of the various classes and subclasses of Igs has its own unique role to play in the immunologic defense of the individual. For example, IgA is the major class of Ig present in all external secretions. It is primarily responsible for protecting mucosal surfaces. IgG subclasses bind Fc receptors differently, and thus vary in effector function.10 Determinants common to subsets of individuals within a species, yet differing between other members of that species, are termed allotypes and define inherited polymorphisms that result from allelic forms of immunoglobulin C (less commonly, V) genes.11
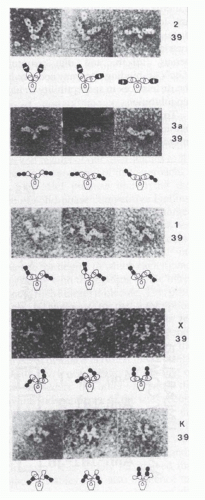 FIG. 5.3. Electron micrographs (top; ×350,000) and interpretive diagrams (below) of murine mAb hybridoma group A carbohydrate (HGAC) 39 (specific for the cell wall polysaccharide of Streptococcus pyogenes) in complex with anti-idiotypic mAb Fab fragments. HGAC 39 is represented in the diagrams as an open figure, and the Fab anti-Id probes are represented as solid figures. The Fab arms of the antibody targets and probes are drawn to indicate their rotational orientation as planar (oval with open center), intermediate (bone shape with or without central opening), or perpendicular (“dumbbell shaped”). Different complexes illustrate the range of Fab-Fab angles made possible by segmental flexibility. 1, anti-IdI-1 Fab; 2, anti-IdI-2 Fab; 39, HGAC 39; 3a, anti-IdI-3a Fab; K, anti-Cκ Fab; X, anti-IdX Fab. IdI designates an individual idiotope, and IdX, a crossreactive idiotope. Antibody complexes were stained with 2% uranyl formate as described by Roux et al.108 Reproduced from Proceedings of the National Academy of Sciences (from Roux et al.108 with permission). |
Glycosylation
N-linked carbohydrates can be found in all constant domains as well as in some variable domains.12 The structure of the attached N-linked carbohydrate can vary greatly, depending on the degree of processing. These carbohydrates can play a major role in Ig function.13 For example, human IgG molecules contain a conserved glycosylation site at Asn 297, which is buried between the CH2 domains.14 This oligosaccharide structure is almost as large as the CH2 domain itself. O-linked sugars are also present in some Igs.12 Human IgA1, but not IgA2, possesses a 13 amino acid hinge region that contains three to five O-linked carbohydrate moieties.15 A deficiency in proper processing of these O-glycans can contribute to IgA nephropathy, which is a disease that is characterized by the presence of IgA1-containing immune complexes in the glomerular mesangium.16
A HISTORICAL PERSPECTIVE
The identification of Ig as a key component of the immune response began in the 19th century. This section describes the history of the identification of Ig and introduces fundamental terminology.
Antibodies and Antigens
Aristotle and his contemporaries attributed disease to an imbalance of the four vital humors: the blood, the phlegm, and the yellow and black biles.17 In 1890, Behring (later, von Behring) and Kitasato reported the existence of an activity in the blood that could neutralize diphtheria toxin.18 They showed that sera containing this humoral antitoxin activity would protect other animals exposed to the same toxin. Ehrlich, who was the first to describe how diphtheria toxin and antitoxin interact,19 made glancing reference to “Antikörper” in a 1891 paper describing discrimination between two immune bodies, or substances.20 The term antigen was first introduced by Deutsch in 1899. He later explained that antigen is a contraction of “Antisomatogen+- Immunkörperbildner,” or that which induces the production of immune bodies (antibodies). Thus, the operational definition of antibody and antigen is a classic tautology.
Gamma Globulins
In 1939, Tiselius and Kabat immunized rabbits with ovalbumin and fractionated the immune serum by electrophoresis into albumin, alpha-goblulin, beta-globulin, and gamma-globulin fractions.21 Absorption of the serum against ovalbumin depleted the gamma-globulin fraction, hence the terms immunoglobulin and IgG. “Sizing” columns were used to separate Igs into those that were “heavy” (IgM), “regular” (IgA, IgE, IgD, IgG), and “light” (light chain dimers). Immunoelectrophoresis subsequently permitted identification of the various Ig classes and subclasses.
Fab and Fc
In 1949, Porter first used papain to digest IgG molecules into two types of fragments, termed Fab and Fc (Table 5.1).22
Papain digested IgG into two Fab fragments, each of which could bind antigen, and a single Fc fragment. Nisonoff developed the use of pepsin to split IgG into an Fc fragment and a single dimeric F(ab)2 that could cross-link antigens.23 Edelman broke disulfide bonds in IgG and was the first to show that IgG consisted of two H and two L chains.24
Papain digested IgG into two Fab fragments, each of which could bind antigen, and a single Fc fragment. Nisonoff developed the use of pepsin to split IgG into an Fc fragment and a single dimeric F(ab)2 that could cross-link antigens.23 Edelman broke disulfide bonds in IgG and was the first to show that IgG consisted of two H and two L chains.24
TABLE 5.1 Definitions of Key Immunoglobulin Structure Nomenclature | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
Two Genes, One Polypeptide
The portion of the constant domain encoded by the Fc fragment was the first to be sequenced and then analyzed at the structural level. It could be readily crystallized when chilled. The heterogeneity of the V domain precluded sequence and crystallographic analysis of an intact Ig chain until Bence-Jones myeloma proteins were identified as clonal, isolated Ig light chains. These intact chains could be purified and obtained in large quantities, which finally permitted rational analysis of antibody structure and function.25 Recognition of the unique nature of a molecule consisting of one extremely variable V domain and one highly conserved C domain led to the then-heretical Dreyer-Bennett proposal of “two genes, one polypeptide,”26 which was subsequently and spectacularly confirmed by Tonegawa.27
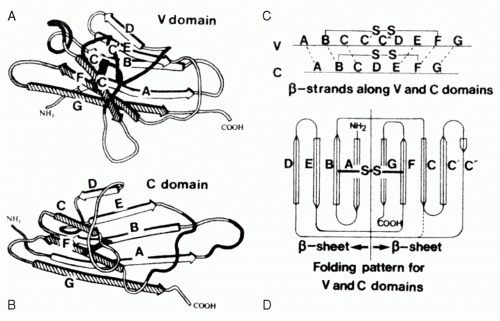 FIG. 5.4. The Immunoglobulin Domain. A: A typical variable domain structure. Note the projection of the C-C′ strands and loop away from the core. B: A typical compact constant domain structure. C: The cysteines used to pin the two b-sheets together are found in the B and F strands. D: The folding pattern for variable and constant domains.2 |
THE IMMUNOGLOBULIN DOMAIN
The Ig domain is the core unit that defines members of the Ig superfamily (reviewed in Williams and Barclay2 and Harpaz and Chothia28). This section describes the Ig domain in detail.
The Immunoglobulin Superfamily
Each Ig domain consists of two sandwiched β-pleated sheets “pinned” together by a disulfide bridge between two conserved cysteine residues (Fig. 5.4). The structure of the β-pleated sheets in an Ig domain varies depending on the number and conformation of strands in each sheet. Two such structures, V and C, are typically found in Igs. C-type domains, which are the most compact, have seven antiparallel strands distributed as three strands in the first sheet and four strands in the second. Each of these strands has been given an alphabetical designation ranging from amino terminal A to carboxy-terminal G. Side chains positioned to lie sandwiched between the two strands tend to be nonpolar in nature. This hydrophobic core helps maintain the stability of the structure to the point that V domains engineered to replace the conserved cysteines with serine residues retain their ability to bind antigen. The residues that populate the external surface of the Ig domain and the residues that form the loops that link strands can vary greatly in sequence. These solvent exposed residues offer multiple targets for docking with other molecules.
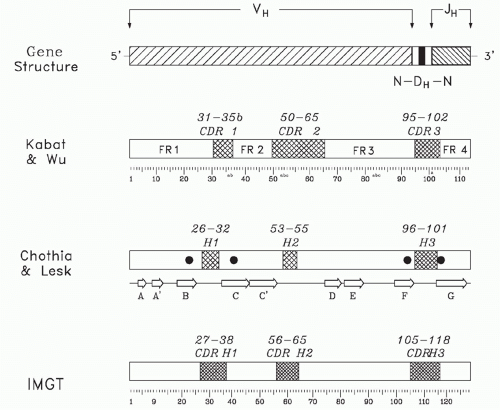 FIG. 5.5. Sequence Conservation and Hypervariability within a Heavy Chain Variable (V) Domain. The primary sequence the V domain can be divided into four regions of sequence conservation, termed framework regions (FRs), and three regions of hypervariability, termed complementarity determining regions (CDRs). A schematic of the genomic origin of the variable domain is shown at the top of the figure. The classic separation of the sequence into FR and CDR by Kabat et al.31 is shown below the gene structure. The letter designation for individual β strands is given beneath the Chothia and Lesk nomenclature,32 which focused more on structure. The positions of each of the four invariant residues of the VH chain (FR1 Cys22, FR2 Trp36, FR3 Cys92, and FR4 Trp103) are shown as darkened circles on the Chothia and Lesk model. The ImMunoGeneTics information system® designation has attempted to rationalize sequence variability with structure and is the current nomenclature of choice.33 |
The V Domain
V-type domains add two additional antiparallel strands to the first sheet, creating a five strand-four strand distribution. Domain stability results from the tight packing of alternately inward-pointing residue side chains enriched for the presence of hydrophobic moieties to create a hydrophobic domain core. The H and L variable domains are held together primarily through noncovalent interaction between the inner faces of the β sheets.29,30
Early comparisons of the primary sequences of the V domains of different antibodies identified four intervals of relative sequence stability, termed framework regions (FRs), which were separated by three hypervariable intervals, termed complementarity determining regions (CDRs) (Figs. 5.5 and 5.6).31 The exact location of these intervals has been adjusted over the years, first by a focus on the primary sequence,31 then by a focus on the three-dimensional structure,32 and, more recently, by a consensus integration of the two approaches by the international ImMunoGeneTics information system® (IMGT)33 (see Fig. 5.5). (For students of the Ig repertoire, IMGT maintains an extremely useful website, http://www.imgt.org, which contains a large database of sequences as well as a multiplicity of software tools.)
The C and C′ strands that define a V domain form FR2. These strands project away from the core of the molecule (see Figs. 5.4 and 5.6) where they take on a conserved structure that is parallel and opposite to the FR2 of the companion V and adjacent to the FR4 of the complementary chain. Approximately 50% of the interdomain contacts in the hydrophobic core of the V domain are formed by contacts between the FR2 of one chain and the FR4 of the complementary chain.30 Another 30% to 45% is contributed by contacts between the CDR3 and the FR2 or CDR3 of the complementary domain. The overall interdomain contact includes between 12 and 21 residues from the L chain V domain and 16 to 22 residues from H chain V domain, most of which are contributed by the FR2, CDR3, and FR4 regions.
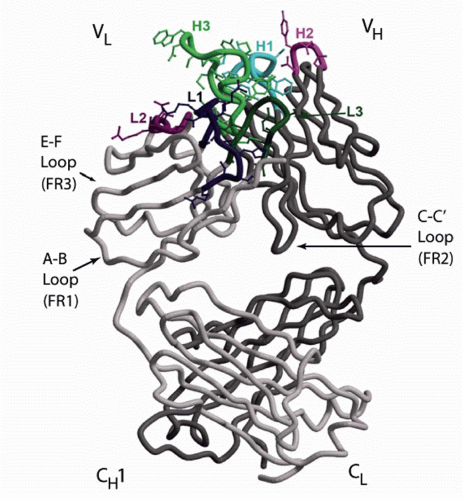 FIG. 5.6. The Structure of an Fab. The antigen-binding site is formed by the heavy (H) and light (L) chain B-C, C′-C″, and F-G loops. Each loop encodes a separate complementarity determining region (CDR). The location of CDRs H1, H2, H3, L1, L2, and L3 are shown. The opposing H and L chain C-C′ strands and loop help stabilize the interaction between VH and VL. This C-C′ structure is encoded by the second V framework region, FR2. The inclusion of this structure permits the variable (V) domains to interact in a head-to-head fashion. The E-F strands and loop are encoded by the FR3 region and lie directly below the antigen-binding site. The A-B strands and loop encode FR1 and lie between the CH1 and CL domains and the rest of the Vs. The beta sheet strands of the CH1 and CL domains rest crosswise to each other. The illustration is modified.203 |
There are approximately 40 crucial sequence sites that influence variable domain inter- and intradomain interactions.32,34 Four of these sites are relatively invariant: the two cysteines that form the disulphide bridge between the beta sheets and two tryptophan (phenylalanine in Jκ) residues, one near the beginning of the C strand and the second near the beginning of the G strand, that pack against the bridge to add stability. Beyond these and other common core residues, Ig domains can vary widely in their primary amino acid sequences. However, a common secondary and tertiary structure characteristic of the core Ig V domain tends to be preserved.
FAB STRUCTURE AND FUNCTION
Introduction
It is the Fab domain that allows Ig to discriminate between antigens. The Fab is individually manufactured to precise specifications by individual developing B cells. It shows an amazing array of binding capabilities while maintaining a highly homologous scaffold. This section describes the characteristics of the Fab domain, its component V domains, and the paratope, which is the part of the Fab that actually binds antigen.
Fab, Fv, and Fb
The antigen-binding fragment (Fab) is a heterodimer that contains an L chain in its entirety and the V and CH1 portions of the H chain (see Figs. 5.1 and 5.6). The Fab in turn can be divided into a variable fragment (Fv) composed of the VH and VL domains, and a constant fragment (Fb) composed of the CL and CH1 domains (see Table 5.1). Single Fv fragments can be produced in the laboratory through genetic engineering techniques.35 They recapitulate the monovalent antigen binding characteristics of the original, parent antibody. Other than minor allotypic differences, the sequences of the constant domains do not vary for a given H chain or L chain isotype. The eponymous V domain, however, is quite variable.
Generation of Immunoglobulin Variable Domains by Recombination
Ig V domain genes are assembled in an ordered fashion by a series of recombination events.27,36,37,38,39 The elegant mechanisms used for the assembly of these genes and the Fvs they create are fully discussed in Chapter 6. However, in order to understand the relationship between antibody structure and function, a brief review is in order.
In BALB/c mice, Ig V assembly begins with the joining of one of 13 diversity (DH) gene segments to one of four joining (JH) gene segments. This is followed by the joining of one of 110 functional variable (VH) gene segments.40,41,42 Each DH gene segment has the potential to rearrange in any one of six reading frames (RFs), three by deletion and three by inversion. Thus, these 127 gene segments can come together in 3.4 x 104 combinations. The numbers of gene segments can vary quite widely between different mouse strains, but the process of assembly and the multiplicative effect of combinatorial diversity is the same.
In the final assembled V domain, the VH gene segment encodes FRs H1 to H3 and CDRs H1 and H2 in their entirety (see Figs. 5.1 and 5.5), and the JH encodes FR-H4. CDR-H3 is created de novo in developing B cells by the joining process. CDR-H3 contains the DH gene segment in its entirety, as well as portions of the VH and JH gene segments.
After a functional H chain has been created, L chain assembly begins. Mice contain two L chain loci, κ and λ. In C57BL/6 mice, the κ locus includes five Jκ gene segments and 140 Vκ gene segments, of which 4 and 73 have been shown to be functional, respectively.43,44,45 This provides 292 combinations. There is only one Cκ.46 The BALB/c λ locus contains three Vλ, three functional Jλ, and two or three functional Cλ chains.47,48 The λ constant domains are functionally indistinguishable from each other. Due to gene organization, the λ repertoire provides at most seven combinations. Each VL encodes FRs L1 to L3, CDR L1 and L2, and two thirds of CDR-L3 (see Fig. 5.1). Each JL encodes one-third of CDR-L3 and FR4 in its entirety. Any one
H chain can combine with any one L chain, thus 211 V, D, and J gene segments can provide approximately 1 × 107 different H:L combinations.
H chain can combine with any one L chain, thus 211 V, D, and J gene segments can provide approximately 1 × 107 different H:L combinations.
At the V→D and D→J junctions, the potential for CDRH3 diversity is amplified by imprecision in the site of joining, allowing exonucleolytic loss as well as palindromic (P junction) gain of terminal VH, DH, or JH germline sequence. B cells that develop after birth express the enzyme terminal deoxynucleotidyl transferase (TdT) during the H chain rearrangement process.27,36 TdT catalyzes the relatively random incorporation of non-germline-encoded nucleotides between VH and DH, and between DH and JH. Each three nucleotides of N addition increase the potential diversity of CDR-H3 20-fold. Thus sequences with nine nucleotides of N addition each between the V→D and D→J junctions would enhance the potential for diversity by (20)6, or by 6 x 107; six-fold greater than the potential diversity provided by VDJ gene segment combinations. These genomic gymnastics permit the length of CDR-H3 to vary from 5 to 20 amino acids among developing B cells in BALB/c bone marrow.49 Together, imprecision in the site of VDJ joining and N addition provides the opportunity to create nearly random CDR-H3 sequence, potentially freeing the CDR-H3 repertoire from germline sequence constraints. Although a limited amount of N addition is observed between VL and JL in human,50,51 N addition in murine L chains is distinctly uncommon. Moreover, the length of CDR-L3 appears to be under relatively strict control, greatly limiting the potential for somatic L chain junctional diversity. 48,50 Thus, CDR-H3 represents the greatest focus for the initial somatic diversification of the antibody repertoire.
Segmental Conservation and Diversity within the V Domain
Although the large numbers of V gene segments might give the impression of a smooth incremental range of available diversity, multigene families are thought to evolve in concert through mechanisms of gene conversion, and V gene segments are no exception. Sequence relationships allow grouping them into families and clans of sequences that share nucleotide homology,52 as well as structural features. Close inspection of the VH gene repertoire has shown that these family relationships reflect segmental gene conversion coupled with selection for function.4,53,54
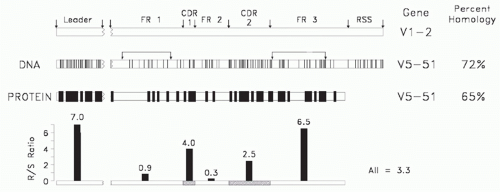 FIG. 5.7. A comparison of two human Clan I VH sequences that belong to different VH families (modified4). Shown is a comparison of the deoxyribonucleic acid and amino acid sequences of the V5-51 and V1-2 gene segments. Each line depicts a divergence in a nucleotide or amino acid at that position. Shown at the bottom is a replacement/silent site substitution analysis by interval. Random mutation tends to exhibit an R/S ratio of 2.9. The smaller the ratio, the greater the preservation of sequence. The intervals identified by the arrows predict the family and clan of origin. |
Due to the need to maintain a common secondary and tertiary core Ig V domain structure capable of associating randomly with a complementary V chain to form a stable Fv, the core sequence of FR2, which is encoded by the VH gene segment, and the core sequence of FR4, which is encoded by the JH gene segment, are highly conserved among all Ig V domains. Conversely, the need to generate a diverse repertoire of antigen-binding sites has led to extensive diversity in the CDR-1 and CDR-2 intervals. One might presume that the FR1 and FR3 intervals, which form the external surface of the antibody, would not be under any particular constraints, but sequence comparisons suggest otherwise.
Given the need to diversify the CDRs and the need to preserve FR2, it is not surprising that family identity, which might reflect ancestral relationships, can be assigned by the extent of FR1 and FR3 similarity.55 Of these, FR1 appears to be under the greatest constraints, with VH gene segments belonging to different families both within and across species barriers exhibiting extensive similarities in FR1 sequence (Fig. 5.7). Sequence similarities in FR1 and, to a lesser extent, FR3 allow grouping of human and murine VH families into three clans of related sequences, presumably reflecting an early divergence in sequence from a primordial VH gene sequence (Fig. 5.8).
Constraints on the Sequence and Structure of Variable-Encoded Complemenatarity Determining Regions
The antigen-binding site of an Ig is formed by the juxtaposition of the six hypervariable H and L chain V domain
intervals: CDRs-H1, -H2, and -H3; and CDRs-L1, -L2, and -L3.31 The CDR sequences of V gene segments tend to be enriched for codons where mutations maximize replacement substitutions.56 This includes the RGYW motif that facilitates somatic hypermutation.57,58 While evolution appears to favor CDR1 and CDR2 sequences that facilitate codon diversity, it also appears to preserve specific loop structures.
intervals: CDRs-H1, -H2, and -H3; and CDRs-L1, -L2, and -L3.31 The CDR sequences of V gene segments tend to be enriched for codons where mutations maximize replacement substitutions.56 This includes the RGYW motif that facilitates somatic hypermutation.57,58 While evolution appears to favor CDR1 and CDR2 sequences that facilitate codon diversity, it also appears to preserve specific loop structures.
 FIG. 5.8. Evolutionary Relationships among Vertebrate VH Families. The sizes or relative placements of the evolutionary connecting lines are not to scale. VH sequences from all mammalian species analyzed to date can be placed into one of these three clans (modified4). |
Although there is great variation in the sequence and size of these CDRs, it has been shown that five of them, CDR-H3 being the notable exception, possess one of a small set of main-chain conformations termed canonical structures.32,34,59,60,61,62 Each canonical structure is determined by the loop size and by the presence of certain residues at key positions in both the loop and framework regions. It can be calculated that the total number of possible combinations of canonical structures, or structure classes, is 300.63 However, only 10 of these combinations, or classes, are sufficient to describe the majority of human and mouse Fab sequences. Among specific classes, the lengths of CDR-H3 and -L1 appear to correlate with the type of recognized antigen. Antibodies with short loops in -H2 and -L1 appear to be preferentially specific for large antigens (proteins), whereas antibodies with long loops in -H2 and -L1 appear to be preferentially specific for small molecules (haptens).63
Given that the sequence and structure of the framework regions, which define families, influences the canonical structure of VH-encoded CDRs, it is not surprising that the structure repertoire of canonical structures is strongly associated with family and clan identity.64 This implies restrictions to the random diversification of the hypervariable loop structures (canonical structures) and their combinations within the same VH gene segment (canonical structure classes). It further suggests evolutionarily and structurally imposed restrictions operating to counteract the random diversification of these CDRs.
Diversity and Constraints on the Sequence and Structure of CDR-H3
The combination of VDJ assortment, variation in the site of gene segment rearrangement, and N nucleotide addition makes CDR-H3 the most variable of the six hypervariable regions. In some cases, the sequence of CDR-H3 appears designed to provide optimal flexibility.65 Correspondingly, it has been more difficult to assign canonical structures to the CDR-H3 loops similar to those observed for the V-encoded CDRs. However, insight into a gradient of possible structures has been gained.
CDR-H3 can be separated into a base, which is adjacent to the frameworks, and a loop. The base tends to be stabilized by two common residues, an arginine at Kabat position 94 (IMGT 106) and an aspartic acid at Kabat 101 (IMGT 116).32,33 These form a salt bridge which, together with the adjacent residues, tends to create one of three backbone conformations, termed kinked, extrakinked, and extended.66 In some sequences with kinked or extrakinked bases, it is possible to predict whether an intact hydrogen-bond ladder may be formed within the loop of the CDR-H3 region, or whether the hydrogen-bond ladder is likely to be broken.67,68 However, for many CDR-H3 sequences, especially those that are longer, current tools provide less than optimal predictions for the structures of individual CDR-H3s.
Despite the potential for totally random sequence provided by the introduction of N nucleotides, close inspection has shown that the distribution of amino acids in the
CDR-H3 loop is enriched for tyrosine and glycine,31,69 and relatively depleted of highly polar (charged) or nonpolar (hydrophobic) amino acids, although the precise pattern depends on the species of origin.70 This pattern of amino acid utilization is established early in B-cell development, prior to the expression of Ig on the surface of the cell (Fig. 5.9)49,71,72,73 and reflects evolutionary conservation of JH and DH gene segment sequences. In particular, although the absolute sequence of the DH is not the same, the pattern of amino acid usage by RF is highly conserved. Of the six potential RFs, RF1 by deletion is enriched for tyrosine and glycine. RF2 and RF3 by deletion are enriched for hydrophobic amino acids, as they are by inversion. RF1 by inversion tends to encode highly polar, often positively charged, amino acids.69 Various species use different mechanisms to bias for use of RF1 by deletion, to limit use of hydrophobic RFs, and to restrict or prevent use of RFs enriched for charged amino acids. Forced rearrangement into RFs with charged amino acids yields an altered repertoire enriched for charge and depleted of tyrosine and glycine.71
CDR-H3 loop is enriched for tyrosine and glycine,31,69 and relatively depleted of highly polar (charged) or nonpolar (hydrophobic) amino acids, although the precise pattern depends on the species of origin.70 This pattern of amino acid utilization is established early in B-cell development, prior to the expression of Ig on the surface of the cell (Fig. 5.9)49,71,72,73 and reflects evolutionary conservation of JH and DH gene segment sequences. In particular, although the absolute sequence of the DH is not the same, the pattern of amino acid usage by RF is highly conserved. Of the six potential RFs, RF1 by deletion is enriched for tyrosine and glycine. RF2 and RF3 by deletion are enriched for hydrophobic amino acids, as they are by inversion. RF1 by inversion tends to encode highly polar, often positively charged, amino acids.69 Various species use different mechanisms to bias for use of RF1 by deletion, to limit use of hydrophobic RFs, and to restrict or prevent use of RFs enriched for charged amino acids. Forced rearrangement into RFs with charged amino acids yields an altered repertoire enriched for charge and depleted of tyrosine and glycine.71
The distribution of CDR-H3 lengths can also be regulated both as a function of differentiation and as a function of ontogeny.49,74 In association with long V-encoded CDRs, short CDR-H3s create an antigen-binding cavity at the center of the antigen-binding site, and CDRs of intermediate length can create an antigen-binding groove. Each species appears to prefer a specific range of CDR-H3 lengths.75 Long CDR-H3s, which can create “knobs” at the center of the antigen-binding site, are unevenly distributed between species and reflect both divergence in germline sequence and somatic selection.
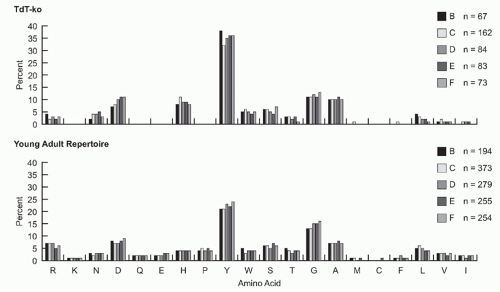 FIG. 5.9. Both in the absence or presence of N-addition, the preference for tyrosine and glycine in complementarity determining region-H3 begins early and intensifies with B-cell development. VH 7183DJCµ transcripts were cloned and isolated from fractions B (pro-B cells) through F (mature B cells) from the bone marrow of 8- to 10-week-old terminal deoxynucleotidyl transferase-sufficient and terminal deoxynucleotidyl transferase-knockout BALB/c mice.73 The amino acids are arranged by relative hydrophobicity, as assessed by a normalized Kyte-Doolittle scale.204,205 Use is reported as the percent of the sequenced population. The number of unique sequences per fraction is shown. |
The Antigen-Binding Site is the Product of a Nested Gradient of Regulated Diversity
The tension between the need to conserve essential structure and the need to emphasize diversity in an environment subject to unpredictable antigen challenge appears to create a gradient of regulated diversity in the Fv. The most highly conserved components of the Fv are FR2 and FR4, which form the hydrophobic core of the VH:VL dimer (see Figs. 5.4 and 5.5). FR1, which in the H chain presents with three conserved structures, helps form the ball and socket joint between the VH and CH1. FR3, which in the H chain defines the family and provides 7 different structures in human versus 16 different structures in mouse, frames the antigen-binding site (Fig. 5.10). The V-encoded CDRs, -H1, -H2, -L1, -L2, and most of -L3, are programmed for diversity. However, conserved residues within these CDRs, which interact with V family-associated FR3 residues, constrain diversity within a
preferred range of canonical structures. CDR-H3, the focus of junctional diversity, lies at the center of the antigen-binding site. The conformation of its base tends to fit within three basic structures. The loop varies greatly in sequence, yet still maintains a bias for the use of tyrosine and glycine. Thus, diversity increases with proximity to the tip of the antigenbinding site but appears to be held within regulated limits.
preferred range of canonical structures. CDR-H3, the focus of junctional diversity, lies at the center of the antigen-binding site. The conformation of its base tends to fit within three basic structures. The loop varies greatly in sequence, yet still maintains a bias for the use of tyrosine and glycine. Thus, diversity increases with proximity to the tip of the antigenbinding site but appears to be held within regulated limits.
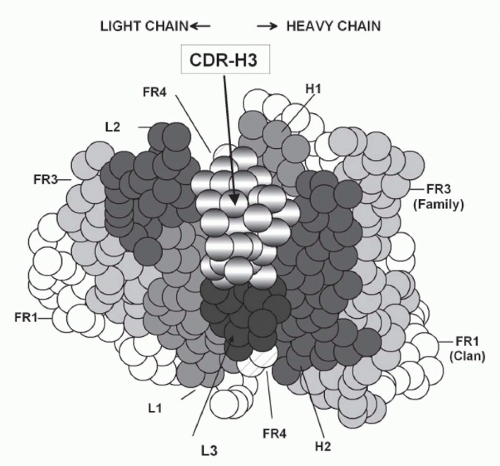 FIG. 5.10. Location and Generation of Complementarity Determining Region-H3. A: A cartoon of the classic antigen-binding site (modified4). Due to its central location, most antigens bound to the antibody will interact with complementarity determining region-H3. |
The extent and pattern of diversity in CDR-H3 can have a critical effect in the biologic function of Ig as a soluble effector molecule. Absence of N addition, with its enhancement of tyrosine and glycine usage (see Fig. 5.9




Related posts:
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
