Immunoglobulins: Molecular Genetics
Edward E. Max
Sebastian Fugmann
INTRODUCTION
To respond to a foreign molecule (antigen) on an invading pathogen, the “humoral” immune system generates antibodies, or immunoglobulins (Igs), that can bind specifically to the offending antigen. Each antibody molecule is composed of two identical light (L) chains and two identical heavy (H) chains, all linked by disulfide bonds to form a symmetric Y-shaped tetramer. The ability of the immune system to generate an antigenspecific antibody depends on the fact that, before exposure to antigen, millions of naïve resting B cells circulate in the individual, each cell displaying on its membrane several thousand identical copies of a single unique species of antibody; these serve as B-cell receptors (BCRs) for that lymphocyte. Only a tiny fraction of the B cells express a BCR capable of binding to any particular antigen. When these B cells bind their antigen, they become activated to proliferate and mature into antibodysecreting plasma cells, which manufacture large amounts of antibody specific for the activating antigen. To be able to generate antibodies against a universe of diverse pathogens, this “clonal selection” mechanism for specific antibody secretion requires an enormous diversity of Ig species expressed on naïve B cells prior to antigen exposure. Indeed, in the 1960s the number of different antibody sequences in the repertoire of typical mouse was estimated in the millions. To encode this many sequences seemed to require an unreasonably high percentage of the mammalian genome (now estimated to contain only about 30,000 genes). Understanding the genetic source of Ig diversity—Ig gene assembly—was the first major challenge and achievement of the molecular biologic investigations of antibody genes, and this will be discussed first in this chapter.
A week or so after antigen administration, the B-cell response changes in two ways that generally improve the protective functions of antibodies. B cells initially express antibodies of the IgM isotype, but cells that migrate into germinal centers receive T-cell-derived stimuli that can induce them to switch to production of IgG, IgA, or IgE without changing their antigen specificity; this switch results from a deoxyribonucleic acid (DNA) recombination event known as class switch recombination (CSR). In addition, over the course of an immune response, the affinity of antibody for antigen gradually improves as a result of somatic hypermutation (SHM) of antibody genes, coupled to selection for B cells expressing high-affinity antibodies. CSR and SHM are discussed later in this chapter.
In this chapter, well-established facts about Ig genes are summarized concisely, while areas currently under investigation are considered in more detail, with particular attention to topics expected to interest immunologists.
OVERVIEW OF IMMUNOGLOBULIN GENE ASSEMBLY
In the 1960s, investigators determined the amino acid sequences of Igs secreted by several mouse myelomas (clonal tumors of B-lymphocytes that secrete a single pure species of Ig). The N-terminal domains of the L and H chains— each roughly 100 amino acids—were highly diverse between different myeloma proteins and were designated variable (V) regions. In contrast, sequences of the remaining domains of the proteins were essentially identical for every myeloma Ig of a given class (and so they were designated constant [C] region domains). The advent of recombinant DNA technology allowed comparisons of V region genes expressed in different myelomas with the corresponding sequences in nonlymphoid DNA (commonly referred to as “germline” DNA). It was found that each myeloma V gene is composed of several segments that are separated in germline DNA; these germline segments must undergo one or more DNA recombination events to assemble a complete V region.1 For example, each complete Vκ gene from a myeloma or B-lymphocyte encodes roughly 108 amino acids and is assembled by linking one of about 40 germline Vκ segments (encoding amino acids 1 through 95) to one (of five) “joining” or Jκ segments encoding residues 96 to 108. Similarly, a complete Vλ gene is assembled from one germline Vλ segment and one Jλ segment. H chains are assembled from three segments; a diversity (D) segment is interposed between VH and JH. In developing B cells, the germline gene segments are assembled into functional V exons by a process named V(D)J recombination (Fig. 6.1).
V(D)J recombination is a “cut and paste” process in which the DNA between two recombining V, D, or J gene segments is excised from the chromosome, and the two remaining DNA segments are joined together to reseal the DNA break. The two principal proteins executing the “cut” phase of this process are encoded by the recombination activating genes (RAG)1 and 2. These proteins recognize unique sequences, known as recombination signal sequences (RSSs), that flank and mark each eligible V, D, and J gene segment (RSSs are described further in the following). After the RAG proteins cut the DNA, the subsequent “joining” of the gene segments relies largely on ubiquitous DNA repair factors.
How Recombination Contributes to Diversity
V(D)J recombination contributes in several distinct ways to the diversity of antigen-binding specificities. First, there is
combinatorial diversity, as each Ig locus contains multiple V, D (in case of the IgH locus), and J segments that can be combined in many ways. The total number of theoretically possible combinations of VH, DH, JH, VL, and JL, is the multiplication product of the numbers of possible H chains— about 40 (VH) × 27 (DH) × 6 (JH) or 6480 combinations in humans—times the number of possible L chain combinations (about 290), or almost 2 million. This repertoire is vastly larger than could be achieved by devoting the same total lengths of DNA sequence to preassembled variable region exons. Second, there is junctional diversity generated by flexibility in the position of joining between gene segments. This was initially recognized by comparisons of nucleotide sequences of various myeloma Vκ genes to their germline Vκ and Jκ precursors. As shown in Figure 6.2A, these comparisons revealed that the crossover point between sequence derived from a germline Vκ region and a Jκ region could vary in different myelomas, increasing the diversity of amino acids around codons 95 and 96. H chain VDJ exons exhibit this flexibility at both V-D and D-J junctions, yielding striking variation in the lengths D regionderived segments, from zero to about 14 amino acids. And additional junctional diversity is produced by the addition of nucleotides not present in any germline elements: “N” and “P” nucleotides, discussed below. Importantly, the three-dimensional structures of Igs established by X-ray crystallography reveal that the VL-JL junction and the VH-DH-JH junction each encode one of the three “complementarity determining region” loops of L or H chain that can contact antigen; thus, this junctional diversity is directly functionally relevant for diversifying antigen binding.
combinatorial diversity, as each Ig locus contains multiple V, D (in case of the IgH locus), and J segments that can be combined in many ways. The total number of theoretically possible combinations of VH, DH, JH, VL, and JL, is the multiplication product of the numbers of possible H chains— about 40 (VH) × 27 (DH) × 6 (JH) or 6480 combinations in humans—times the number of possible L chain combinations (about 290), or almost 2 million. This repertoire is vastly larger than could be achieved by devoting the same total lengths of DNA sequence to preassembled variable region exons. Second, there is junctional diversity generated by flexibility in the position of joining between gene segments. This was initially recognized by comparisons of nucleotide sequences of various myeloma Vκ genes to their germline Vκ and Jκ precursors. As shown in Figure 6.2A, these comparisons revealed that the crossover point between sequence derived from a germline Vκ region and a Jκ region could vary in different myelomas, increasing the diversity of amino acids around codons 95 and 96. H chain VDJ exons exhibit this flexibility at both V-D and D-J junctions, yielding striking variation in the lengths D regionderived segments, from zero to about 14 amino acids. And additional junctional diversity is produced by the addition of nucleotides not present in any germline elements: “N” and “P” nucleotides, discussed below. Importantly, the three-dimensional structures of Igs established by X-ray crystallography reveal that the VL-JL junction and the VH-DH-JH junction each encode one of the three “complementarity determining region” loops of L or H chain that can contact antigen; thus, this junctional diversity is directly functionally relevant for diversifying antigen binding.
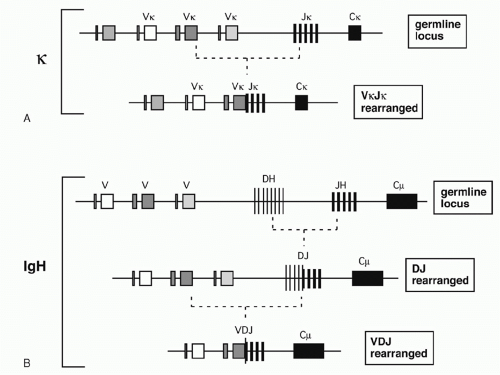 FIG. 6.1. Variable Assembly Recombination. A: In the κ locus, a single recombination event joins a germline Vκ region with one of the Jκ segments. B: In the immunoglobulin H locus, an initial recombination joins a diversity segment to a JH segment. A second recombination completes the variable assembly by joining a VH to DJH. |
The imprecision of V(D)J recombination increases Ig diversity, but at a cost. Because the precise boundaries between V, D, and J result from independent stochastic events, only about one-third of all recombination events maintain the correct reading frame through the J segments. Gene rearrangements leading to functional Ig genes are often referred to as “productive,” while out-of-frame rearrangements are labeled “nonproductive.”
Function of Recombination Signal Sequences
Analysis of DNA sequences flanking the germline V, D, and J gene segments revealed highly similar sequence motifs that have subsequently been shown to define targets for V(D)J recombination: the RSSs, which serve as the recognition sequences for the V(D)J recombinase proteins RAG1 and RAG2, as mentioned previously. Notably, RSSs lie adjacent to L- and H-chain Ig gene segments and to T-cell-receptor (TCR) gene elements throughout phylogeny. RSSs consist of a conserved seven base pairs (bps) long “heptamer” (consensus: CACAGTG) and a nine bp long “nonamer” sequences
(consensus: ACAAAAACC) that are separated by less wellconserved spacers of either approximately 12 or 23 bp in length (Fig. 6.3). Based on the spacer lengths, the two classes of RSSs are referred to as 12-RSSs and 23-RSSs, respectively. (Note that some laboratories use the term recombination signal instead of RSS in their publications.)
(consensus: ACAAAAACC) that are separated by less wellconserved spacers of either approximately 12 or 23 bp in length (Fig. 6.3). Based on the spacer lengths, the two classes of RSSs are referred to as 12-RSSs and 23-RSSs, respectively. (Note that some laboratories use the term recombination signal instead of RSS in their publications.)
 FIG. 6.2. Vκ-Jκ Recombination at Single Base Resolution. A: The sequence of the recombined MOPC41 κ gene around the VJ junction is shown (center) with the sequences of the two germline precursors (Vκ41 and Jκ1) shown above and below. The germline origins of the recombined gene are indicated by the vertical lines and the shading of the V-derived sequence. B: The consequences of joining the same germline sequences (from part A) at four different positions are shown. Of the four alternative recombination products illustrated, the top one is that actually found in MOPC41. The second example has a single nucleotide difference but no change in encoded amino acid sequence. The third and fourth alternatives yield Arg or Pro at position 96; both of these amino acids have been found at this position in sequenced mouse κ chains. |
Recombination occurs almost exclusively between coding sequences associated with RSSs of different spacer lengths, a requirement referred to as the “12/23-rule” (i.e., the recombination between two 12-RSSs [or two 23-RSSs] is “forbidden” and does not occur in vivo). Within each gene locus, all gene segments of one class (e.g., all Vs in the Igκlocus) carry RSSs with the same spacer length. Thus the 12/23 rule drives appropriate recombination events leading to functional VJ and VDJ products, and prevents futile recombination events, such as between two V or two J gene segments. While the heptamer and nonamer are the major determinants of RSS function necessary for V(D)J recombination, increasing evidence suggests that spacer sequences can modulate recombination efficiencies of compatible gene segments (e.g., they affect the non-random usage of human Vκ elements2).
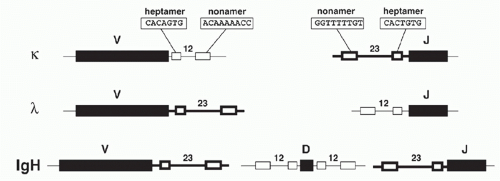 FIG. 6.3. Conserved Elements Flank Germline Variable (V), Diversity (D), and Joining (J) Region Genes. Conserved heptamer and nonamer recombination signal sequences (RSSs) lie adjacent to V, D, and J coding sequences and are important for targeting V(D)J recombination. The heptamer and nonamer elements are separated by spacer regions of about 12 basepairs (bp) (thin lines) or 23 bp (thick lines). Depending on the locus, V regions may be flanked by 12 bp or 23 bp RSS; and similarly for J regions. But one of each type of element must be present for recombination to occur, a requirement that prevents futile recombination events (e.g., J to J). |
THE THREE IMMUNOGLOBULIN GENE LOCI
To understand the contribution of the germline V, D, J element repertoire to Ig diversity, several laboratories undertook cloning and sequence analysis of individual V region genes from the IgH, Igκ, and Igλ loci of human and mouse. More recently, the complete sequences of all human and mouse Ig loci have been determined as part of the genome sequencing projects for these two species (available online at www.ncbi.nlm.nih.gov, though annotation that describes
function and refers to earlier literature is incomplete). It is important to point out that Ig gene loci are not identical between individuals (humans) or between individual strains of inbred mice. Several Internet resources are devoted to providing convenient updated access to Ig germline gene sequences. The international ImMunoGeneTics database (http://imgt.org) includes a database for Ig and TCR genes from a variety of species, and includes maps, sequences, lists of chromosomal translocations, and multiple helpful links. IgBLAST (www.ncbi.nlm.nih.gov/igblast/) is a service of the National Center for Biotechnology Information and allows a submitted sequence to be searched against known annotated germline V, D, and J sequences.
function and refers to earlier literature is incomplete). It is important to point out that Ig gene loci are not identical between individuals (humans) or between individual strains of inbred mice. Several Internet resources are devoted to providing convenient updated access to Ig germline gene sequences. The international ImMunoGeneTics database (http://imgt.org) includes a database for Ig and TCR genes from a variety of species, and includes maps, sequences, lists of chromosomal translocations, and multiple helpful links. IgBLAST (www.ncbi.nlm.nih.gov/igblast/) is a service of the National Center for Biotechnology Information and allows a submitted sequence to be searched against known annotated germline V, D, and J sequences.
TABLE 6.1 Overview of the Number of Variable, Diversity, and Joining Segments in Each of the Three Immunoglobulin Loci in Humans and Mice | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The Murine Immunoglobulin H Germline Variable, Diversity, and Joining Gene Segments
VH Segments
The murine VH region extends over about 2.5 megabases on chromosome 12 and includes roughly 100 functional segments (depending on mouse strain) plus additional VH pseudogene segments (Table 6.1). All VH elements are in the same transcriptional orientation as the D, JH, and CH regions.3 The VH segments are classified into 16 distinct families based on sequence similarity; VH elements within a family show more than 80% nucleotide sequence identity. Elements of individual VH families are largely clustered together, though some interdigitation occurs (Fig. 6.4). The VH families can be grouped into three “clans” based on sequence conservation primarily of their framework regions (framework region 1, codons 6 to 24, and framework region 3, codons 67 to 85, respectively), which form the more conserved structural backbone of the Ig variable region. Importantly, these clans are conserved between man, mouse, and frog, suggesting that their emergence in the repertoire preceded the amphibian-reptile divergence.4
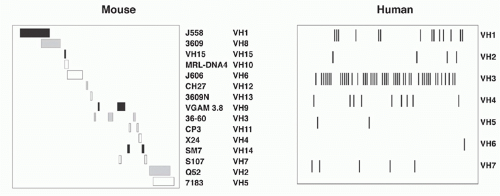 FIG. 6.4. Maps of the Murine and Human VH Loci. The 15 known murine VH gene families are shown in their approximate map positions. Each rectangle represents a cluster of VH genes of the indicated family; the clan identification of the VH families is indicated by the color of the rectangle: black for clan I, gray for clan II, and white for clan III. Although some interdigitation is shown by overlapping families (e.g., the Q52 and 7183 families), the murine VH families are largely clustered. In contrast, all human VH genes (vertical lines) of a prototypic haplotype are shown in the right panel; extensive interdigitation of families is apparent. |
Diversity and JH
About 50 kb downstream of the most 3′ VH element resides the murine D cluster spanning about 80 kb, depending on
the mouse strain (see Table 6.1). Each D segment is flanked by 12 RSSs on both sides, so that the 12/23 rule ensures that all assembled V genes carry a D element between their V and J segments (which are both flanked by 23 RSSs that prevent direct V to J rearrangements).
the mouse strain (see Table 6.1). Each D segment is flanked by 12 RSSs on both sides, so that the 12/23 rule ensures that all assembled V genes carry a D element between their V and J segments (which are both flanked by 23 RSSs that prevent direct V to J rearrangements).
The murine D elements are classified into four families: DSP2, DFL16, DST4, and DQ52. Although D regions could theoretically contribute to Ig diversity by being read in all three frames, the mouse has evolved mechanisms that strongly favor one of them.5 Four functional germline JH sequences reside about 0.7 kb downstream of the most 3′ D region, DQ52.
The Human Immunoglobulin H Germline Variable, Diversity, and Joining Gene Segments
VH Segments
The human VH locus spans 1.1 Mb at the telomeric end of chromosome 14 (14q32.33) (see Table 6.1). The human germline VH segments—numbering roughly 40 to 45—fall into seven families that, in contrast to the family clusters characteristic of the murine locus, are extensively interdigitated (see Fig. 6.4). Some human VH sequences are polymorphic owing to VH insertions or deletions in different allelic chromosomes. Twenty-four additional germline VH sequences have been mapped to chromosome 15 and 16 and represent nonfunctional “orphans” that were apparently duplicated from the IgH locus on chromosome 14.6
Diversity and JH Regions
Twenty-six human D elements are located in an ˜40 kb region about 20 kb downstream of VH6, the most 3′ of the VH genes.7 This D cluster is comprised of four tandem duplications of a 9.5 kb segment containing a representative of each of six D families. The twenty-seventh D element—DHQ52— is the only one showing sequence similarity to a mouse segment (DQ52) and shares a homologous location just 5′ to JH1. In contrast to mice, humans use all reading frames of D elements.7 One reading frame encodes primarily hydrophilic residues, one encodes hydrophobic residues, and one includes frequent stop codons. Some D elements contain stop codons that can be removed by nuclease trimming during VDJ assembly. As in mice, the human JH cluster is immediately downstream of DHQ52.
Heavy Chain Constant Regions
Murine and human genomic clones containing C region H-chain (CH) genes include separate exons encoding the ˜100 to 110 amino acid Ig domains. These domains were independently identified by internal homologies of amino acid sequences and by three-dimensional structural analysis (X-ray crystallography). The exons are separated from each other by introns of roughly 0.1 to 0.3 kb. Thus, for example, the mouse γ2b protein has three major domains (CH1, CH2, and CH3) with a small hinge domain between CH1 and CH2. The gene structure may be summarized as follows:
CH1 – intron – hinge – intron – CH2 – intron – CH3
(292) (314) (64) (106) (328) (119) (322)
where the numbers in parentheses represent the number of nucleotides in each segment. As an interesting contrast, the hinge region of the α gene is encoded contiguously with the CH2 domain with no intervening intron, while the unusually long human γ3 hinge is encoded by three or four hinge exons.
Genomic Organization of the CH Region
Each B-lymphocyte initially produces IgM by expressing an assembled variable region linked to Cµ, but may use CSR (discussed later in this chapter) to replace Cµ with one of the several CH regions lying downstream, thereby allowing expression of IgG, IgA, or IgE (Fig. 6.5A). Eight murine CH genes span about 200 kb of DNA on chromosome 12; these genes were linked by contiguous clones in 1982.8 Several γ pseudogenes lie within the clustered γ functional genes9 (Fig. 6.5B). The coding sequences of all CH genes are oriented in the same direction.
The human CH genes were similarly cloned, and then eventually completely linked by the Human Genome Project. The human IgH locus contains a large duplication, with two copies of a γ-γ-ε-α unit separated by a γ pseudogene (see Fig. 6.5B). One of the duplicated ε sequences is also a pseudogene, and a third closely homologous ε-related sequence—a “processed” pseudogene—is present on chromosome 9.
The IgH locus has also been examined in several other species besides mouse and human, and several notable differences have been observed. Rabbits, for example, have 13 Cα sequences and only a single Cγ gene10; this unusual expansion of genes contributing to mucosal immunity may be related to the peculiar habit of coprophagy in these animals. In contrast to the multiplicity of rabbit Cα genes, pigs have only one Cα gene and eight Cγ genes. Camels are unusual in having H chains that function in the absence of L chains.11
Membrane versus Secreted Immunoglobulin
Igs are found either as secreted molecules in the serum or as membrane-bound receptors. The membrane-bound µ chains contain a C-terminal hydrophobic transmembrane domain consisting of 26 uncharged hydrophobic amino acids encoded by additional membrane exons, and these residues anchor the protein in the cell membrane lipid bilayer. The membrane (µm) and secreted (µs) forms are derived from the same gene by alternative splicing (Fig. 6.6). The same general gene structure has been found for other CH genes, suggesting that differential splicing accounts for the two forms of all Ig isotypes.
Early B cells make roughly similar quantities of both µm and µs, whereas maturation to the plasma cell stage is associated with strong predominance of µs production, facilitating high-level secretion of circulating Ig. The balance between the two ribonucleic acid (RNA) splice forms of µ has been interpreted as a competition between splicing of the CH4 and M1 exons versus the cleavage/polyadenylation at the upstream µs poly(A) site. These processes are mutually exclusive because CH4-M1 splice removes the µs poly(A) site, while cleavage at the µs poly(A) site removes the membrane exons.
Cis-regulatory elements (and corresponding transacting RNA binding proteins) control the balance between these
processes. They include a GU-rich element downstream of the µs poly(A) site,12 the polyadenylation factor cleavage stimulator factor 64,13 and the U1A protein.14 These factors likely function downstream of B-lymphocyte-induced maturation protein-1 (BLIMP-1) whose expression in Ig-secreting plasma cells was also found to be critical for µs poly(A) site utilization.15 Cis-acting sequences affecting the ratio of alternative splice forms have been described for other isotypes besides Cµ, particularly Cα.16
processes. They include a GU-rich element downstream of the µs poly(A) site,12 the polyadenylation factor cleavage stimulator factor 64,13 and the U1A protein.14 These factors likely function downstream of B-lymphocyte-induced maturation protein-1 (BLIMP-1) whose expression in Ig-secreting plasma cells was also found to be critical for µs poly(A) site utilization.15 Cis-acting sequences affecting the ratio of alternative splice forms have been described for other isotypes besides Cµ, particularly Cα.16
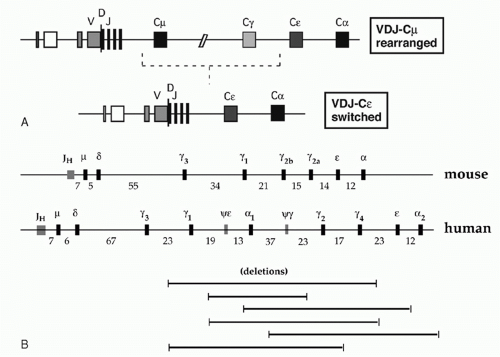 FIG. 6.5. Deletional Isotype Switch Recombination. A: The expression of “downstream” heavy chain genes is accomplished by a recombination event that replaces the Cµ gene with the appropriate heavy chain constant gene (Cε is shown as an example), deleting the deoxyribonucleic acid between the recombination breakpoints. B: The murine and human heavy chain constant region genes are diagrammed with the approximate intergene distance indicated below (in kb); various literature values for these distances differ somewhat, possibly due to allelic polymorphisms. The human locus shows a large duplication of γγεα sequences. |
Membrane Ig serves as the antigen-recognition component of the BCR that is critical for initiating the signal for lymphocyte activation following contact with antigen. Transduction is mediated by an associated protein dimer composed of the BCR components Igα and Igβ (CD79a and CD79b) whose cytoplasmic domains contain immunoreceptor tyrosine-based activation motifs similar to those found in the CD3 chains mediating TCR signaling. Additional signaling is mediated by conserved tyrosines in the cytoplasmic tails of the IgG and IgE H chains, which serve as a phosphorylation-dependent docking sites for the signaling adapter Grb2.17 Binding of Grb2 enhances BCR signaling and subsequent B-cell proliferation.
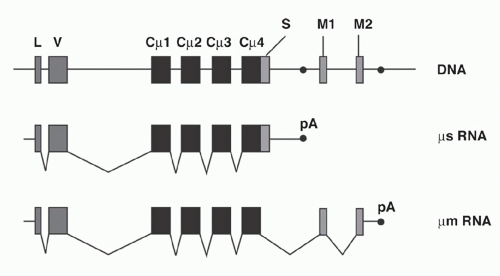 FIG. 6.6. Two Ribonucleic Acids (RNAs) Generated from the µ Gene by Alternative Processing. The top line illustrates the exons of the µ gene (black rectangles) in an expressed, rearranged µ gene. A primary transcript including all the exons present in the deoxyribonucleic acid can be processed as shown to yield either µs RNA (containing a C-terminal “secreted” sequence) or µm RNA (containing the two membrane exons). |
Kappa Light Chain Genes
Murine Germline Vκ Locus
The murine Vκ locus spans about 3.2 mb on chromosome 618 and contains 20 Vκ families, some of which are shared by human and mouse (see Table 6.1). Vκ sequences within a single family are largely clustered together. Some Vκ elements lie in the opposite orientation to that of the Jκ and Cκ elements, and these Vκ segments undergo VJ recombination by an inversion rather than deletion (Fig. 6.7). A few Vκ sequences have been localized to chromosome 16 and 19 and are considered orphan genes.
Human Germline Vκ Locus
The human Vκ locus (see Table 6.1) lies on the short arm of chromosome 2 (2p11-2) spanning ˜2 mb of DNA.19 The locus includes a large inverted duplication, so that most Vκ sequences exist in pairs with one copy lying in the cluster proximal to Jκ (and in the same orientation) and a second copy (inverted) in the distal cluster. The average sequence similarity between duplicates is 98.9%, suggesting the duplication occurred less than 5 million years ago. This is consistent with the absence of such duplication in chimpanzees, which diverged from the human lineage approximately 6 million years ago. Interestingly, about 5% of human alleles also lack the distal duplication.
Outside the Igκ locus, at least 25 orphan Vκ segments have been identified in clusters on chromosome 1, 2, and 22. The orphan cluster located in the long arm of chromosome 2 was probably separated from the major locus—on the short arm of this chromosome—by a pericentric inversion (which must have occurred rather recently in evolution as it is absent from chimpanzee and gorilla).
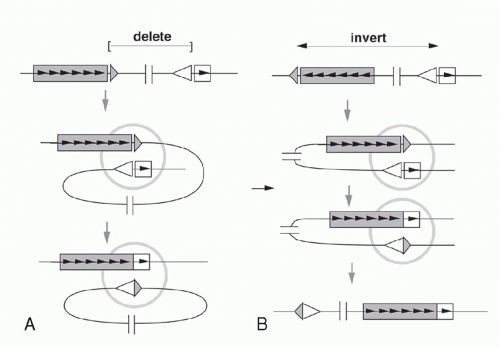 FIG. 6.7. The Same “Micro” Mechanism of Recombination can Join Vκ and Jκ by Deletion or Inversion, Depending on the Relative Orientation of the Two Precursors in Germline DNA. A: When V coding sequence (shaded rectangle) and J coding sequence (white rectangle) are oriented in the same 5′ → 3′ direction in germline DNA (as indicated by the internal arrowheads), the recombination yields a VJ coding joint plus a DNA circle containing the signal joint (apposed triangles). B: If V is oriented in the opposite direction in germline DNA, an identical recombination reaction at the “micro” level (inside shaded circle) leaves the signal joint linked to the recombined VJ coding joint. |
Jκ and Cκ Elements
In comparison to the H chain genes, the organization of the C region segments in the κ locus is relatively simple (see Table 6.1). A single Cκ gene with a single exon and with no reported alternative splice products is found in both mouse and human. While all five Jκ elements are functional in humans, the third J element in mice has not been observed in functional κ L chains.
Apart from the typical Vκ-Jκ rearrangements, an additional recombination event occurs uniquely in the κ locus. The event is mediated by V(D)J recombination utilizing a 23-RSS element—designated Recombining Sequence in the mouse20 and Kappa Deleting Element in the human21—that is positioned in an intergenic region downstream of Cκ; the recombination results in the deletion of the Cκ exon. Hence, Cκ fragments are undetectable on Southern blots of DNA from λ-expressing human lymphoid cells,22 as in most B cells the Cκ genes are apparently deleted from both chromosomes before Igλ gene rearrangement begins.
Lambda Light Chain Genes
Murine λ Locus
In laboratory mouse strains, only about 5% of the B-lymphocytes utilize Igλ L chain, and the diversity of these L chains is meager due to the very small number of V region genes (Fig. 6.8). Complete sequence analysis23 of the murine locus revealed two V-J-C clusters (Vλ2-Vλx-Jλ2Cλ2-Jλ4Cλ4 and Vλ1-Jλ3Cλ3-Jλ1Cλ1) separated by about 110 kb. Each Jλ is linked to its own Cλ region gene, but Jλ4 is nonfunctional. Recombination occurs largely within each cluster, although Vλ2Cλ1 products are occasionally observed. The ancestry of the Vλx element is uncertain, as it is rather dissimilar to the other Vλ segments; indeed, it resembles Vκ as much as Vλ. In contrast to the Igκ locus, the Vλ segments are flanked by 23-RSS and the Jλ gene segments by 12-RSS (see Fig 6.3).
Human λ Locus
The human Vλ region was characterized by intensive cloning, sequencing, and mapping of Vλ elements and ultimately by the complete sequence analysis of 1 Mb covering the entire locus24 (see Table 6.1). Within the Vλ cluster lies the human VpreB gene (discussed below), as well as several genes and pseudogenes unrelated to the Igλ system.
λ L chains are much more abundant in man than in mouse (about 40% of human L chains are λ versus about 5% in mouse). Four forms of human λ chains have been classified serologically, with differences residing in a small number of amino acids in the C region. The serologic classification of Kern+ corresponds to a glycine at position 152 versus a serine in Kern-. The Oz+ designation corresponds to a lysine at position 190 versus an arginine in the Oz- variant. Similarly, Mcg+ λ chains (versus Mcg-) contain asparagine 112 (versus alanine), threonine 114 (versus serine) and lysine 163 (versus threonine).
Four functional human Jλ-Cλ segments and three pseudogenes are clustered within an approximately 33 kb region of DNA (see Fig. 6.8) and the four major expressed human λ isotypes correspond to the functional JCλ1, JCλ2, JCλ3, and
JCλ7, with the latter encoding an isotype provisionally designated Mcp.25 JCλ6 may be functional in some individuals, and the common allele—which has a 4 bp insertion leading to a deletion of the C-terminal third of the Cλ region— can nevertheless undergo Vλ-Jλ recombination, encoding a truncated protein that can associate with H chains. A variety of polymorphic variants of the human λ locus have been detected, apparently the result of gene duplication, as shown in Figure 6.8.26 Lastly, three Cλ-related sequences have been discovered near the major Jλ-Cλ cluster. One of these, designated λ14.1, represents the human homolog of the murine “surrogate” L chain λ5 (see following discussion).
JCλ7, with the latter encoding an isotype provisionally designated Mcp.25 JCλ6 may be functional in some individuals, and the common allele—which has a 4 bp insertion leading to a deletion of the C-terminal third of the Cλ region— can nevertheless undergo Vλ-Jλ recombination, encoding a truncated protein that can associate with H chains. A variety of polymorphic variants of the human λ locus have been detected, apparently the result of gene duplication, as shown in Figure 6.8.26 Lastly, three Cλ-related sequences have been discovered near the major Jλ-Cλ cluster. One of these, designated λ14.1, represents the human homolog of the murine “surrogate” L chain λ5 (see following discussion).
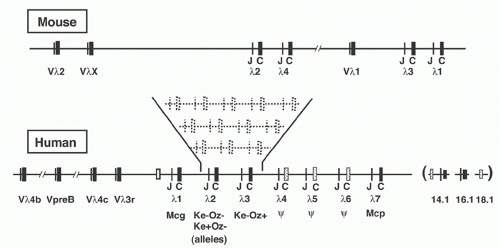 FIG. 6.8. Germline λ Genes. The maps in this figure are schematic (i.e., not to scale). A: The murine λ gene system includes four JC complexes and three V genes, as shown. B: The human λ locus includes multiple V genes, of which only three are shown. The human VpreB “surrogate” light chain gene is located within the Vλ cluster. The Cλ locus includes a segment of seven JC complexes plus three additional unlinked sequences. The hatched JC complexes diagrammed above the seven linked λ sequences represent polymorphic variants with additional duplications of the JC unit. The 14.1 sequence—the human λ5 “surrogate” light chain homolog—lies downstream of the JC cluster. Exon 1 of the 14.1 gene is homologous to an exon upstream of Jλ1 (as indicated by the unlabeled white rectangle). |
λ-Related “Surrogate” Light Chains
Ig H chains cannot reach the cell surface without pairing with Ig L chains. However, Ig µ H chains can be detected on the surface of pre-B cells whose Igκ and Igλ loci are still in their germline configuration and thus do not produce L chains. In these cells, a “surrogate L chain” (SLC) composed of two smaller proteins, VpreB and λ5, facilitates the surface expression of the µ H chain protein. The first component (λ5) was identified as the product of a gene expressed exclusively in pre-B cells that showed high sequence similarity to the J and C regions of the λ locus,27 As four murine Cλ genes were already known, it was designated λ5. The second component of the SLC was identified as a gene residing about 4.7 kb upstream of λ5 in the mouse genome. Based on its similarities to both Vλ and Vκ (and its expression in pre-B cells), it was called VpreB1. A second, nearly identical sequence in the mouse genome is named VpreB2 and appears to be functional,28 and a less similar VpreB3 has also been described. Neither λ5 nor VpreB genes show evidence of gene rearrangement in B or pre-B cells, and homologs have been found in every mammalian species examined.
The two SLC proteins form a L chain-like heterodimer that is able to fulfill some functions of a true L chain, including association with µ H chains to permit surface µ expression prior to the availability of κ or λ L chains. Thus, when a µ H chain gene was transfected into an Ig-negative myeloma line, no surface µ expression was observed unless λ5 and VpreB genes were also transfected.29 Surface µ chains are covalently linked to the λ5 protein, while the VpreB1 protein is noncovalently associated. The expression of µ-SLC on the surface of pre-B cells triggers the onset of Vκ-Jκ rearrangement, as discussed below.
V(D)J RECOMBINATION
The mechanism by which germline variable region segments (VL and JL, or VH, D, and JH) are assembled in the DNA to form a complete active V region has been pursued ever since Ig gene recombination was first discovered. In this section we will address 1) the molecular mechanism of the reaction, 2) the topology of the recombination events, 3) the components of the recombinase machinery, and 4) the regulation of that machinery during B-cell development.
Molecular Mechanism of V(D)J Recombination
Recombination Model Overview
A model for the detailed mechanism of the V(D)J recombination event must account for the observed features of the recombination products and of their germline precursors. In the germline precursors, the RSSs with their heptamer, nonamer, and appropriate 12 or 23 bp spacers are necessary and sufficient to create efficient recombination targets; model substrates in which RSSs flank DNA sequences completely unrelated to Ig genes are competent to undergo recombination. The model shown in Figure 6.9 will serve as a framework for discussion of the recombination mechanism. The recombination is thought to begin with binding of the RAG1-RAG2 complex to the RSSs that flank the two gene segments to be recombined. Simultaneous DNA cleavage occurs precisely between the RSSs and the gene segments. The two ends of the RSSs (frequently named “signal ends”) are joined directly, forming “signal joints.” In contrast, the ends of the gene segments (also referred to as “coding ends”) are processed prior to joining and are ultimately ligated together, giving rise to “coding joints” and completing the recombination event.
Recombination Products: Coding Joints and Signal Joints
In the recombination products, signal joints are typically direct ligation products of the signal ends: the RSSs are joined directly at the heptamers (“back-to-back”), and nucleotide additions or deletions at these junctions are quite rare. The properties of the coding joints, however, are more complex, as the joining reaction at these DNA ends is “imprecise.” The following features are frequently present:
Deletions: variable number of bases are deleted from the ends of the coding regions (in comparison to the “complete” sequence in the germline precursor)
Nongermline (“N”) nucleotides: random nucleotides (with a bias toward G and C) are added by a templateindependent DNA polymerase (discussed below). The sequence of these N nucleotides has no relationship to the germline V, D, or J sequences.
Palindromic (“P”) nucleotides: the ends of the coding gene segments are sealed by a DNA hairpin structure (see Fig. 6.9, and discussed in the following). “Opening” of these hairpins frequently occurs by nicking at some distance away from the hairpin tip leading to single-stranded overhangs. Filling in of such overhangs by DNA polymerases generates DNA palindromes that mirror the nucleotides at the end of the V, D, or J segment.31 P nucleotides are generally only one or two bps, but they can be longer, especially in mice with the severe combined immunodeficiency defect (SCID) disorder in which the opening of the hairpins occurs in an aberrant manner.
Recombination Intermediates: Blunt Signal Ends and Hairpin Coding Ends
To study broken DNA ends as intermediates in V(D)J recombination, several laboratories employed ligation-mediatedpolymerase chain reaction (LM-PCR) to detect signal ends. This technique involves ligating blunt double-stranded oligonucleotide linkers to blunt genomic DNA breaks, and then amplifying the ligation junctions between a primer in the ligated oligonucleotide and a primer based on known sequence from the ligated genomic DNA; amplification products can
then be cloned and sequenced. LM-PCR analyses of both TCR and Ig genes undergoing V(D)J recombination showed the signal ends to be blunt double strand breaks (dsbs), usually exactly at the heptamer border.32 Similar LM-PCR experiments failed to detect the coding ends unless they were pretreated with mung bean nuclease, a single-strandspecific endonuclease that recognizes the distortion of DNA at a hairpin structure. Sequences of these LM-PCR products from coding ends suggested that the hairpins are precisely at the end of the coding elements, usually without loss or gain of a single nucleotide.33 By Southern blot analyses, coding ends were found to have two properties suggestive of a hairpin-like structure: 1) resistance to exonuclease treatment, and 2) doubling of the apparent length of restriction fragments under denaturing electrophoresis conditions.34
then be cloned and sequenced. LM-PCR analyses of both TCR and Ig genes undergoing V(D)J recombination showed the signal ends to be blunt double strand breaks (dsbs), usually exactly at the heptamer border.32 Similar LM-PCR experiments failed to detect the coding ends unless they were pretreated with mung bean nuclease, a single-strandspecific endonuclease that recognizes the distortion of DNA at a hairpin structure. Sequences of these LM-PCR products from coding ends suggested that the hairpins are precisely at the end of the coding elements, usually without loss or gain of a single nucleotide.33 By Southern blot analyses, coding ends were found to have two properties suggestive of a hairpin-like structure: 1) resistance to exonuclease treatment, and 2) doubling of the apparent length of restriction fragments under denaturing electrophoresis conditions.34
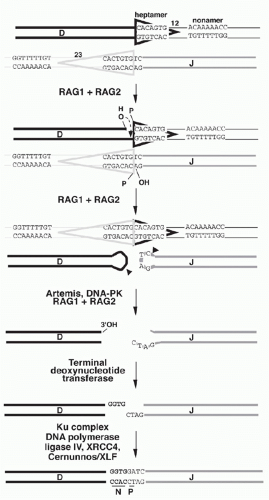 FIG. 6.9. Model for V Assembly Recombinations. All V assembly recombination reactions (in immunoglobulin and T-cell receptor genes) may proceed by a common mechanism, illustrated here by D-J recombination. The recombination signal sequences (RSSs) are included in triangles, which is the conventionally used RSS graphic. Hairpin loops are created on coding ends dependent on the action of the two Recombination Activating Genes: RAG1 and RAG2. After the opening of the hairpin loops, the pictured diversity coding sequence shows the effects of “nibbling” by exonuclease, but the joining coding sequence is spared and shows P nucleotide generation; N region addition is pictured in this example as occurring only on the diversity region end. In reality, exonuclease digestion and N nucleotide addition can occur on either (or both) ends. The steps in the proposed mechanism are discussed in the text. |
Hairpin ends represent V(D)J recombination intermediates that, in wild-type cells, are opened at the hairpin tip (or a few nucleotides away from it) by the Artemis nuclease (discussed below). P nucleotides result from opening the loop at an asymmetric position (see Fig. 6.9); this model would explain why P nucleotides are never observed at coding ends that have been “nibbled” after opening of the hairpin. P nucleotide segments in the rare coding joints observed in SCID mice are unusually long and likely result from resolution of hairpins by nicking enzymes that, unlike Artemis, do not focus on the area near the tip of hairpin loops but instead nick in variable positions in the double-stranded hairpin “stem.”34
Topology of V(D)J Recombination
Deletion versus Inversion
If a V segment and a J segment are both oriented in the same direction, they can recombine by excising the DNA between the coding sequences and ligating the two coding ends. Ligation of the two signal ends produces a DNA circle that generally lacks replication origins and therefore fails to replicate as cells divide after V(D)J recombination. Such excision circles are therefore generally absent in mature B-lymphocytes that have already undergone several rounds of proliferation after completing the Ig gene assembly. By isolating circular DNA from cells actively undergoing Vκ-Jκ rearrangement, it is possible to isolate and characterize the circular molecules bearing signal joints.35
As mentioned previously, some germline Vκ genes are oriented in the opposite direction from the Jκ-Cκ region. In these cases, VJ recombination occurs by an inversion of the DNA between the recombining V and J segments, leaving both the VκJκ coding joint and the signal joint (formed by ligating the RSSs) retained in the chromosome (see Fig. 6.7). This demonstrates that the enzymatic machinery “sees” only the DNA in the immediate vicinity of the recombination site and is insensitive to the topology of the DNA strands far from this site.
Nonstandard Joints
In addition to the canonical coding and signal joints, several “nonstandard” recombination joints have been documented, that, though not contributing to physiologic Ig gene assembly, represent tell-tale signs of a recombination event.36 In the first phase of V(D)J recombination, the DNA is cut at both gene segment-RSS boundaries that participate in the reaction, thereby generating four DNA ends. In principle, there are three possible topologies in which these DNA ends can be rejoined:
“Signal and coding joints”: the standard reaction product in which the two coding ends get joined generating the assembled VJ gene and the 12-RSS/23-RSS signal joint.
“Open and shut joints”: the RSSs get ligated back to the gene segments from which they were released. These joints are topologically identical to the starting DNAs, but can be distinguished from them if nucleotides have been added or deleted at the junctions.
“Hybrid joints”: joints in which the RSSs have traded places so that the 23-RSS that was flanking the Vκ segment is now linked to the Jκ segment, and vice versa.
Secondary V(D)J Recombination
As discussed previously, imprecise joining of gene segments causes about two-thirds of all recombination products to be out-of-frame. Thus, a B-lymphocyte could end up with nonproductively rearranged Igκ genes on both alleles. However, germline Vκ segments lying upstream of an initial Vκ-Jκ recombination junction can recombine with Jκ segments lying downstream of the junction, producing a “secondary” recombination event, as shown in Figure 6.10A.
Such secondary recombination also occurs in cells that have assembled a productive Vκ-Jκ joint if the encoded antigen binding domain recognizes an autoantigen. This type of secondary recombination, known as “receptor editing,” is considered in more detail later in this chapter.
Such secondary recombination also occurs in cells that have assembled a productive Vκ-Jκ joint if the encoded antigen binding domain recognizes an autoantigen. This type of secondary recombination, known as “receptor editing,” is considered in more detail later in this chapter.
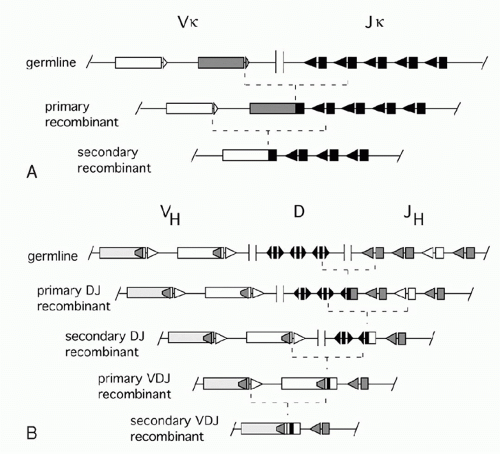 FIG. 6.10. Secondary Recombinations. A: In the κ light chain system, a primary recombination can be followed by recombination between an upstream V and a downstream J. B: Analogous secondary recombinations can occur in the heavy chain system between upstream D and downstream J segments. After V(D)J recombination eliminates all 12-RSS signal elements from the chromosome, secondary recombination can still occur between VH (23-RSS) and an internal heptamer within the VH coding sequence of the VDJ unit. |
In the IgH locus, secondary D-J rearrangements sometimes occur, but only until VH-DJH recombination removes all unused upstream DH segments (Fig. 6.10B). VDJ rearrangement eliminates all the 12-RSSs from the IgH locus that could pair with the 23-RSSs flanking the upstream VH elements. Sometimes, these VH segments do, however, recombine with an established VDJ unit, displacing most of the originally assembled VH element,38 a process sometimes called VH replacement. Such events are mediated by cryptic RSSs (mainly a heptamer sequence) that is present near the 3′ end of about 70% of all VH genes (see Fig. 6.10B). Such internal cryptic RSSs are not generally found in L chain genes. As discussed previously for the L chain, secondary recombination represents a rescue mechanism for cells with nonproductive rearrangements on both H chain chromosomes, and for cells whose encoded antibody recognizes an autoantigen.
The V(D)J Machinery
Since the discovery of V(D)J recombination as the process that assembles the germline antigen receptor gene segments into functional genes, one major question was the identity of the enzymatic machinery catalyzing this complex set of reactions. Genetic and biochemical work by a large number of laboratories led to identification of a total of 13 different proteins that have been shown to be directly involved in V(D)J recombination: RAG1, RAG2, HMG1, Ku70, Ku80, DNA-PKcs, Artemis, pol µ, pol λ, TdT, XRCC4, Cernunnos/XRCC4-like factor (XLF), and DNA ligase IV. The only lymphoid-specific factors are RAG1, RAG2, and TdT; all others are ubiquitously expressed in all cell types, and this feature allows investigators to study aspects of V(D)J recombination by ectopically expressing the RAG proteins in nonlymphoid cells. A recent biochemical tour de force study showed that coding joint formation could be recapitulated in vitro using artificial recombination substrates and highly purified preparations of all 13 proteins.39 The respective coding joints showed all of the features typically observed in vivo (nucleotide deletion, N nucleotide, and P nucleotide addition), suggesting that most, if not all, of the factors involved in the coding end processing steps of V(D)J recombination have been identified. In contrast, signal joint formation was not observed. This step seems to require the removal of the RAG proteins after the cleavage reaction and is likely to require additional factors as yet unidentified.
Recombination Activating Gene Proteins: Mediators of Early Steps in V(D)J Recombination
A major advance in the investigation of V(D)J recombination was the identification of two genes whose products are critical for this process in the B and T cell lineages. In the pioneering experiments, Schatz and Baltimore40 stably transfected fibroblasts with a construct containing a selectable marker whose expression was dependent on V(D)J recombination; as expected, no measurable recombination occurred in this nonlymphoid cell. However, when either human or murine genomic DNA was transfected into these fibroblasts, a small fraction of recipient cells stably expressed recombinase activity, activating the selectable marker. This suggested that a single transfected genomic DNA fragment was able confer recombinase activity in a fibroblast. (Presumably the fibroblast contained endogenous copies of the same genes, but their expression was repressed by mechanisms that could not repress the transfected genes.) This active fragment was cloned and turned out to contain two closely linked genes, designated RAG1 and RAG2, respectively. Both RAG1 and RAG2 are essential for recombination; therefore, these genes would not have been discovered by this transfection technique if they had not been closely linked in the genome. The genes are notable for having no introns splitting up their open reading frame in most species, and for their opposite transcriptional orientation in all species examined.
A crucial role for the RAG genes in V(D)J recombination was supported by the conservation of these genes in all jawed vertebrate species analyzed thus far, from shark through man. RAG1 and RAG2 are expressed together in developing B and T cells, specifically at the stages at which V(D)J recombinase activity is required for the assembly of Ig and TCR genes. Moreover, mouse strains in which either gene has been eliminated by homologous recombination (gene “knockouts”) have no mature B or T cells, as the result of their inability to initiate V(D)J recombination.41,42 Similarly, a subset of human patients with SCID syndrome characterized by the complete absence of T- or B-lymphocytes have been found to have null mutations in RAG genes.43 Patients with hypomorphic alleles often have a complex set of features (oligoclonal T cells, hepatosplenomegaly, eosinophilia, decreased serum Ig but elevated IgE) known as the Omenn syndrome, which can also be caused by defects in other genes involved in V(D)J recombination. Interestingly, the same RAG mutation in different patients can cause either Omenn syndrome or SCID, depending on unknown factors.44
RAG1 shows intrinsic binding affinity for the RSS nonamer sequence via its nonamer binding domain even in the absence of RAG2. Exhaustive mutational analysis has revealed that RAG1 contains the catalytic center of the RAG complex, composed of three amino acids critical for all enzymatic activity: D600, D708, and E962.45,46 RAG2, on the other hand, serves as a regulatory cofactor; it has no intrinsic binding affinity for RSSs, but once bound to RAG1 improves the strength and specificity of RAG1 RSS contacts.47,48 It is also enhances RAG activity on chromosomal substrates and it restricts V(D)J recombination to the G0/G1 stage of the cell cycle (both features are discussed below).
Attempts to determine the molecular role of the RAG proteins in cell-free recombination assays were initially hampered by poor solubility of the proteins, but functional analyses of truncated RAG genes (using RAG expression vectors cotransfected into fibroblasts along with recombination substrate plasmids) revealed that surprisingly large segments of both proteins could be deleted without eliminating recombinase activity, and some of the remaining core regions were
soluble and could be handled relatively easily in experiments. This work allowed the demonstration that in a cell-free in vitro system, core regions of the two RAG proteins together are capable of carrying out cleavage of substrate DNAs as well as hairpin formation on the coding end.49
soluble and could be handled relatively easily in experiments. This work allowed the demonstration that in a cell-free in vitro system, core regions of the two RAG proteins together are capable of carrying out cleavage of substrate DNAs as well as hairpin formation on the coding end.49
The RAG-mediated cleavage occurs in two steps: first a nick is introduced on the top strand between a gene segment and the adjacent heptamer (see Fig. 6.9), then the 3′-hydroxyl group participates as the nucleophile in a direct transesterification reaction to attack the phosphodiester bond adjacent to the heptamer on the bottom strand (see Fig. 6.9), yielding a DNA hairpin structure on the coding end and a new 3′-hydroxyl group on the 3′ end of the bottom heptamer strand.50 After DNA cleavage, the RAG proteins remain in a complex with the DNA ends and facilitate aspects of the joining phase. Mutant forms of RAG1 or RAG2 have been reported that are competent for cleavage but show impairment in coding or signal joint formation.51
While nicking can occur asynchronously at the 12-RSS and 23-RSS, hairpin formation is “coupled” and occurs synchronously at both RSSs. In vitro, coupled cleavage requires only the RAG proteins, HMG1/2 (discussed below) and Mg2+ as the divalent metal ion in the reaction buffer. In vivo, DNA dsb formation at an individual RSS is dangerous as it could give rise to translocations, and it is thought that Mg2+ promotes an optimal molecular “architecture” for controlled V(D)J recombination. In vivo experiments indeed suggest that RAG proteins may bind to and introduce a nick at a single 12-RSS, but do not complete DNA cleavage until a matching 23-RSS is captured into the RAG-RSS complex.52
In addition to the “classical” activities of RAG proteins on DNA segments containing RSSs, these proteins can also catalyze DNA strand cleavage on “nonstandard” substrates.
Transposition. In vitro, purified recombinant core RAG proteins can catalyze the excision and insertion of a DNA fragment with signal ends into foreign DNA, acting as a transposase.53,54 This property provides additional support for the early speculation that the V(D)J recombination system may have originated by insertion of transposon-like DNA fragment encoding RAG genes (and bearing RSSs at its ends) into a primordial antigen receptor gene, thereby generating a pair of separated V and J gene segments. This model of the origin of V(D)J recombination is consistent with the many mechanistic similarities at the molecular level between Ig gene rearrangements and transposition,55 and the recent identification of the Transib transposase family that shows striking sequence similarity to RAG1 and is widespread in insect, echinoderm, helminth, coelenterate, and fungal genomes.56 The recent finding of an apparent homolog of the entire RAG1 and RAG2 gene locus in a sea urchin genome suggests that the two RAG genes may have entered the genome of a common ancestor of all deuterostomes far earlier than the Ig-/TCR-based adaptive immune system developed.57 It remains unclear whether the primordial RAG transposon encoded solely RAG1 (which would then have integrated next to the primordial RAG2 gene) or both RAG1 and RAG2. The transposase activity of RAGs, however, seems to be almost completely suppressed in vivo, and the C-terminus of RAG2 may have evolved to control this potentially deleterious activity.51,58,59,60
VH replacement. As mentioned previously, recombination events can occur between a VH 23-RSS and cryptic RSS within rearranged VH coding sequences. An in vitro model suggests that in VH replacement, the RAG proteins nick both DNA strands without forming a hairpin coding end.61 Whether this is indeed a completely different activity is unclear.
Translocations at non-RSS sequences. The RAG complex also generates two nicks to cleave within the major breakpoint region of the Bcl2 gene. This 150-bp segment is the target of a common RAG-catalyzed translocation between the IgH locus and the Bcl2 gene occurring in most follicular lymphomas. In this segment, there are no RSSs, and the RAG proteins recognize an unusual sequence-dependent DNA conformation different from the normal B-form double helix.62
Although the “core” RAG proteins have been useful for elucidating the molecular mechanism of the cleavage step of V(D)J recombination in biochemical studies, it is clear that the “noncore” portions of each protein confer important functions, as expected from their sequence conservation across species. Broadly speaking, the “noncore” regions ensure regulated and efficient recombination on the physiological substrates (i.e., imperfect RSSs deviating from the perfect consensus heptamer and nonamer) in the context of chromatin. The functions of the “noncore” regions have largely been inferred by comparing V(D)J recombination products from cells expressing core RAG proteins versus full-length versions, and more recently by in vitro studies using full-length RAG proteins that are now available for such analyses.
The C-terminal region of RAG2 has multiple functions and is important for achieving normal numbers of B- and T-lymphocytes in vivo,63 for the formation of precise signal joints during IgH recombination,64 and for protecting against RAG-mediated DNA transposition.51,65 These functions are thought to be conferred at least in part, by a plant homeo domain (PHD) zinc finger fold that is formed by amino acids 414 to 487 in murine RAG2. This PHD domain binds specifically to the tails of histone H3 that are trimethylated at lysine 4 (H3K4Me3),66,67,68 a histone modification that is associated with “open” chromatin and that is uniquely present on “accessible” RSSs in Ig loci (discussed below). In vitro studies suggest that the binding of the RAG2 PHD domain to histone tails causes a conformational change that increases the catalytic activity of the RAG complex.69
Furthermore, the RAG2 C terminus regulates RAG2 protein levels—and hence V(D)J recombinase activity—across the cell cycle to prevent dsbs during DNA synthesis or mitosis, when such breaks could lead to chromosomal deletions.32 RAG1 protein and messenger RNA (mRNA) transcript levels of both RAG genes vary little across the cell cycle, but phosphorylation of RAG2 at Thr490 by the cyclin-dependent
kinase cdk2 mediates its destruction via ubiquitination and proteasomal degradation during S phase.70 Mice expressing RAG2 with a T490A mutation (which cannot be phosphorylated) showed RAG2 protein and dsbs throughout the cell cycle, demonstrating the importance of the RAG2 degradation signal in cell-cycle control of V(D)J recombination.71,72
kinase cdk2 mediates its destruction via ubiquitination and proteasomal degradation during S phase.70 Mice expressing RAG2 with a T490A mutation (which cannot be phosphorylated) showed RAG2 protein and dsbs throughout the cell cycle, demonstrating the importance of the RAG2 degradation signal in cell-cycle control of V(D)J recombination.71,72
The N-terminal noncore region of RAG1 is required in vivo for optimal RAG1 activity and for the formation of precise signal joints in D-J recombination.64 This region of RAG1 contains a RING finger domain that seems to be required for ubiquitination of several proteins, including histone H3.73
Apart from the obvious importance of the RAG proteins in understanding the initial steps of V(D)J recombination, knowledge of these proteins and their genes has allowed two major technical advances that have opened the way to many additional experiments. First, various nonlymphoid cell lines with known defects in various DNA repair genes have been transfected with the RAG genes to identify genes involved V(D)J recombination (these factors are described below). Second, availability of the RAG1 and RAG2 knockout mice has been instrumental in a large number of immunology studies. These mice completely lack functional B cells or T cells, and are not “leaky” like SCID mice, which develop some functional B and T cells, especially as the animals age. Thus the RAG-deficient mice can be used to study the importance of the “innate” immune system (i.e., responses that occur in the absence of antigen-specific lymphocytes) in particular immune responses. They can also be used as recipients for various lymphocyte populations to explore the roles of different cell types. They can also be used as recipients for various lymphocyte populations to explore the roles of different cell types. They can be transfected with transgenes encoding specific Ig genes to study the roles of specific antibodies in B cell development and in immune responses. Finally, they can be used in “RAG complementation” experiments designed to assess the phenotype —in lymphocytes—of various other gene knockouts.74 In RAG complementation, embryonic stem cells in which the gene of interest has been knocked out by homologous recombination are injected into homozygous RAG2 knockout (RAG2-/-) blastocysts. This procedure yields chimeric mice in which all B and T cells derive from the embryonic stem cells deleted for the gene of interest, as these are the only source of intact RAG genes to support lymphocyte development. Such animals can be made more easily than a knockout mouse line, and can be used to study the effect of gene deletion in lymphocytes independent of effects the deletion may have in other cells. In particular, for cases where the gene knockout causes embryonic lethality due to effects on nonlymphoid cells, RAG complementation allows the selective knockout in lymphocytes to be studied in the background normal gene expression in nonlymphoid cells.
High Mobility Group Proteins
The search for RAG cofactors that stimulate cleavage activity in biochemical assays led to the identification of HMG1.75 HMG1 (and the closely related HMG2) are abundant and ubiquitous proteins that bind DNA in a non-sequence-specific manner and to cause a local bend in DNA. The two RAG proteins can form a stable signal complex with a 12-RSS, but efficient complex formation with a 23-RSS requires the addition of either HMG1 or HMG2.76 HMG1/2 apparently stabilizes the bending of the 23-RSS that is induced by the RAG proteins themselves.77
Nonhomologous End Joining Components
The RAG proteins are the essential lymphocyte-specific factors in the DNA cleavage phase of V(D)J recombination, but DNA repair factors that are part of a DNA repair pathway known as nonhomologous end joining (NHEJ) are essential for the joining phase. NHEJ is the major pathway for repair of dsbs (such as those induced by ionizing radiation or reactive oxygen species) during the G0-G1 phases of the cell cycle. (In the S and G2 phases, the additional chromatid genome copy enables breaks to be repaired by homologous recombination.) The six classical core components of NHEJ are Ku70, Ku80, DNA-PKcs, XRCC4, DNA Ligase IV, Artemis, and Cernnunos/XLF, but additional proteins play a role in some models of NHEJ.
The DNA-PK Complex. The first gene for an NHEJ component to be recognized as participating in V(D)J recombination was the SCID gene. This gene was originally identified as being mutated in the scid mouse strain that is immunodeficient due to a marked impairment in V(D)J recombination of both Ig and TCR genes. Lymphocytes from scid mice are able to perform the RAG-mediated cleavage reaction, and can also form signal joints, but are markedly defective in coding joint formation. Subsequently, it was found that the scid mutation also impairs NHEJ, causing radiosensitivity.
The gene mutated in the scid mouse strain encodes DNAPKcs, a large protein (460 kD) with a kinase domain near its C terminus that is related to phosophoinositide-3-kinase (PI3K). This kinase is DNA-dependent and represents the catalytic subunit (hence “cs”) of a heterotrimer known as the DNA-PK complex. The other components are Ku70 and Ku80 (also referred to as Ku86), which were originally identified as the autoantigens recognized by a patient antiserum (Ku was the coded name of the patient, and the numbers refer to the approximate size of the proteins, 70 kD and 80 to 86 kD, respectively). Together, these two very abundant proteins form a heterodimer that binds to the ends of double-stranded DNA independent of the nucleotide sequence of the DNA. The DNA-Ku complex can then recruit DNAPKcs and activate autophosphorylation of this protein.78 In vitro activation of DNA-PKcs was found to be efficient when DNA ends either were at high concentration or, if at low concentration, were on DNA fragments long enough to circularize readily. In contrast, when the DNA-PKcs was located on the ends of DNA fragments too short to circularize (and too dilute for efficient intermolecular interactions with other DNA ends), the DNA-PKcs activation was much reduced. These observations suggest that kinase activation can occur only after two DNA ends are brought together by DNA-PKcs in “synapsis.”79,80 Further phosphorylation of
DNA-PKcs inactivates the protein and may prepare it for removal once DNA ends have been sealed.
DNA-PKcs inactivates the protein and may prepare it for removal once DNA ends have been sealed.
Ku genes are highly conserved through evolution, and homologs are even found encoded in the genome of some bacteria, consistent with a function in general NHEJ not restricted to V(D)J recombination. While mice with a targeted deletion of DNA-PKcs resemble the original scid mutation (i.e., defective coding but functional signal joint formation81,82), Ku70 and Ku80 mutant cell lines are defective in both signal and coding joint formation, and Ku70- and Ku80-deficient mice exhibit a complete block in B- and T-cell development due to their inability to undergo V(D)J recombination.83,84,85
DNA Ligase IV and XRCC4. An important role of activated Ku-DNA-PKcs complex is to recruit the additional components of NHEJ. One such component is DNA ligase IV, which is recruited to the Ku complex and activated by the protein XRCC4.86,87 The evidence suggests that DNA ligase IV is the essential ligase that joins DNA ends in V(D)J recombination and NHEJ. Human patients with ligase IV deficiency (characterized by hypomorphic alleles) have a severe phenotype including chromosomal instability, developmental and growth retardation, radiosensitivity, and immunodeficiency with a T-B-NK+ phenotype.88 The rare DH-JH junctions detected show extensive nucleotide deletion consistent with delayed ligation and prolonged exonuclease digestion.89 In mice, disruption of either the XRCC4 or the DNA ligase IV gene causes embryonic lethality associated with neuronal apoptosis. Crossing these mice with p53 mutants does not improve V(D)J recombination, but rescues the mice from embryonic lethality, suggesting that neuronal cells may be unusually susceptible to p53-triggered apoptosis induced by normal low-level DNA damage during brain development; a similar mechanism may explain the severe human phenotype.90 DNA ligase IV is the only NHEJ component absolutely required to join compatible sticky DNA ends in vitro, though XRCC4 can stimulate this activity significantly.87
Cernunnos/XRCC4-like Factor. The next NHEJ component was independently discovered by two laboratories. One group used yeast two-hybrid screening to search for proteins interacting with XRCC4.91 The other group searched for the gene causing a syndrome of T+ B lymphocytopenia, increased radiosensitivity, and microcephaly in a Turkish family; these investigators used functional cDNA rescue of a patient’s cell line from a radiomimetic drug to identify the gene.92 The protein identified by both groups is a 299 amino acid nuclear protein, which was named Cernunnos or XLF. The protein has a predicted secondary structure similar to that of XRCC4, to which it binds in cells93 as expected from its isolation via two-hybrid screen. When Cernunnos/XLF-deficient fibroblasts were transfected with RAG genes and a recombination substrate, imprecise signal joining was observed, similar to the defect in patients with hypomorphic DNA ligase IV mutations. These experiments all suggest a role for Cernunnos/XLF linked to the function of XRCC4 and ligase IV.
Artemis. The coding ends generated by RAG cleavage cannot be directly ligated because of their hairpin structure, and therefore V(D)J recombination requires a single-strand endonuclease activity to cleave the hairpins. This activity is conferred by the protein named Artemis, which was discovered through positional cloning of the genetic defect in a group of human SCID patients with defects in V(D)J recombination and increased radiation sensitivity.94 Patients with homozygous null mutations of Artemis survive (no embryonic lethality) and show sensitivity to γ irradiation as well as defects in coding joints, while signal joint formation is normal. Hypomorphic Artemis mutations can cause features of the Omenn syndrome similar to those observed with hypomorphic RAG gene mutations.95 Purified recombinant Artemis protein has an intrinsic exonuclease activity in vitro; however, when complexed with DNA-PKcs in the presence of DNA ends, it gains a single-strand endonuclease activity and, in an ATP-dependent step, becomes phosphorylated at multiple sites in the C-terminal region of the protein.96,97 The Artemis endonuclease can cleave synthetic and RAG-generated hairpin ends as well as other singlestranded DNA near a transition to double-strand DNA.98
DNA Polymerase X Family Members. If a hairpin opening leaves blunt ends or complementary sticky ends (like the ends generated by many restriction enzymes), in vitro joining experiments suggest that these ends can be joined by ligase IV without any additional processing.99 However, as Artemis probably opens most hairpins noncomplementary DNA overhangs, further processing of DNA ends generally occurs before ligation completes the recombination. This processing may include further nuclease digestion (by Artemis or exonucleases) and apparently also involves variable DNA extension by three DNA polymerases—polymerase λ, polymerase µ, and terminal deoxynucleotidyl transferase (TdT)—all of which are members of the polymerase X family. Interestingly, all three proteins contain a Brca1-C-terminus domain, which is thought to confer binding to Ku.100
Terminal Deoxynucleotidyl Transferase and N Regions. TdT, the primary source of untemplated “N region” additions in VDJ junctions, is an enzyme uniquely expressed in the thymus and bone marrow; in the B lineage, it is expressed almost exclusively in pro-B cells. It catalyzes the nontemplated addition of nucleotides to the 3′ end of DNA strands. Though no template determines the nucleotides added, the enzyme adds dG residues preferentially, consistent with N region sequences observed in VDJ joints. Both TdT expression and N nucleotide addition are characteristically absent from fetal lymphocytes.101 N region addition is common in H chain genes (recombined in pro-B cells) but rare in murine L chain genes (recombined in pre-B cells), though perhaps somewhat less rare in human.102 This is consistent with the observation that in mice the expression of a µ H chain may downregulate TdT expression,103 contributing to the reduced level during the stage of L chain recombination.
Lymphocytes with engineered defects in their TdT genes produced rearranged Ig V regions with almost no N additions. Conversely, when TdT expression was engineered in cells undergoing κ or λ L chain rearrangement, the level of
N nucleotide addition to these coding joints was dramatically increased. Furthermore, mice engineered to undergo premature Vκ-Jκ joining in pro-B cells show an increased frequency of N region nucleotides in their recombined Vκ genes.104 These results suggest that the low frequency of N region sequences in normal κ or λ recombinations is caused by the reduced levels of TdT at this stage of B-cell development (see following discussion).
N nucleotide addition to these coding joints was dramatically increased. Furthermore, mice engineered to undergo premature Vκ-Jκ joining in pro-B cells show an increased frequency of N region nucleotides in their recombined Vκ genes.104 These results suggest that the low frequency of N region sequences in normal κ or λ recombinations is caused by the reduced levels of TdT at this stage of B-cell development (see following discussion).
The absence of N region addition in TdT mutant mice, as well as in normal fetal lymphocytes, is associated with an increase in the frequency of recombination junctions with microhomologies. These are short stretches of nucleotides that are present close to the end of both germline gene segments involved in the recombination event. These junctions suggest a joining intermediate in which the complementary single-stranded regions from the two coding ends hybridize to each other, much as “sticky ends” generated by restriction endonucleases can facilitate ligation of DNA fragments. This alternative joining pathway may restrict the diversity of neonatal antibodies; the resulting antibodies are possibly enriched in specificities for commonly encountered pathogens, or have broadened specificity, as has been reported for TCRs lacking N regions.105 Decreased N region nucleotides and a high incidence of homology-mediated recombination have also been found in the rare coding joints formed in Ku80-/- mice, consistent with a role for Ku in recruiting TdT or supporting its action.106
Polymerase µ and Polymerase λ. Polymerase µ and polymerase λ are ubiquitously expressed polymerases. Both readily fill in single-strand gaps in DNA and apparently participate in V(D)J recombination by filling in single-strand 3′ overhangs generated by asymmetric hairpin opening. Without this filling in, such overhangs might be resected by nucleases. Indeed, when in vitro NHEJ reconstitution experiments are performed using purified proteins and DNA fragments with overhanging ends, the omission of polymerase µ or polymerase λ increases the deletional trimming at junctions.100 Similar excessive deletions at VDJ junctions are observed in mice lacking polymerase µ or polymerase λ. Remarkably, however, polymerase µ knockout mice show abnormalities only in their L chains,107 whereas the deletions in polymerase λ knockouts are restricted to their H chains.108 This selectivity may be explained by corresponding changes in the relative mRNA levels for these two polymerases at different stages of B-cell development.
Other Participants in V(D)J Recombination
DNA Damage Response Factors. In eukaryotic cells, DNA breaks initiate signals that halt cell division, induce DNA repair, and in some cases trigger apoptosis. Several proteins apart from NHEJ components can be detected at DNA breaks induced by V(D)J recombination or irradiation, including γ-H2AX, a phosphorylated form of the histone H2AX; ATM, the product of the gene mutated in the disease ataxia telangiectasia; Nbs1 (or nibrin), the product of the gene mutated in Nijmegen breakage syndrome; and 53BP1, p53 binding protein 1. The importance of these proteins in V(D)J recombination is not clear because defects in all three are compatible with near normal V(D)J recombination. Possibly, they participate in backup mechanisms to prevent aberrant V(D)J recombination and thus translocations.
Pax5/B-Cell-Specific Activator Protein. Pax5 (also known as B-cell-specific activator protein; BSAP) is a transcription factor required for normal B-cell development. Pax5-deficient mice are able to complete DJH recombination, but VH to DJH recombination is impaired except for certain VH genes located proximal to the D regions. Interestingly, 94% of human and mouse VH coding genes were found to have potential Pax5 binding sites. Surprisingly, Pax5 was found to coimmunoprecipitate with RAG proteins, to potentiate in vitro cleavage of a VH gene RSS, and to enhance VH to DJH recombination in RAG-transfected fibroblasts; the latter enhancement required intact Pax5 binding sites in the VH sequence.109
REGULATION OF V(D)J RECOMBINATION IN B-CELL DEVELOPMENT
The expression of only one antigen binding specificity by each B-lymphocyte is a crucial requirement of the clonal selection model of the humoral immune response. Thus, the recombination events that occur between Ig gene segments are carefully regulated so that most B cells express only one L chain isotype, either Igκ or Igλ (isotype exclusion), and use only one of the two alleles of H and L chain genes (allelic exclusion). These constraints ensure that each B cell expresses a single H2L2 combination. Current evidence suggests that V(D)J recombination is controlled largely at two levels: regulation of the RAG protein activity and regulation of accessibility of the germline V, D, and J elements to the recombinase machinery. Both of these are controlled by the stage of B-cell development; conversely, the expression of Ig provides a signal critical for regulating maturation of B cells. A brief scheme of B-cell development is presented in the following as background.
B- and T-lymphocytes differentiate from pluripotent hematopoietic stem cells in the fetal liver and bone marrow (Fig. 6.11). The primordial lymphoid progenitor has the potential to differentiate into B- or T-lymphocytes or natural killer cells. Among the earliest markers that indicate B-lineage specificity are the non-Ig components of the pre-BCR: Igα, Igβ, and λ5. CD19, which functions as a coreceptor in signal transduction, first appears in large proliferating “pro-B” cells, which also express several other distinguishing surface markers including c-kit, B220, TdT, and CD43. RAG gene expression in pro-B cells initiates D to J rearrangements on both alleles. Subsequently, recombination with germline VH elements occurs; if the recombination is “productive” (i.e., yielding an “in-frame” VDJ junction), a µ H chain protein can be produced. This protein appears on the B-cell surface along with SLC in a pre-BCR (also named µ-SLC) complex that also includes Igα and Igβ. As the resulting large pre-B cells proliferate, RAG gene expression declines. After several rounds of division, the cells become smaller, stop dividing, turn up RAG gene expression once more, undergo L chain recombination, and express surface
IgM. These “immature B cells” again turn down RAG expression. In these IgM+IgD- immature B cells, contact with autoantigens may upregulate RAG expression again to facilitate receptor editing (discussed in more detail below). When immature B cells eventually also express surface IgD, they become “mature B cells” and migrate into the periphery, ready to be triggered by antigen exposure.
IgM. These “immature B cells” again turn down RAG expression. In these IgM+IgD- immature B cells, contact with autoantigens may upregulate RAG expression again to facilitate receptor editing (discussed in more detail below). When immature B cells eventually also express surface IgD, they become “mature B cells” and migrate into the periphery, ready to be triggered by antigen exposure.
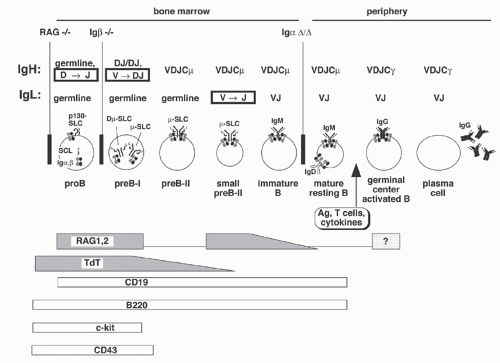 FIG. 6.11. Immunoglobulin (Ig) Gene Recombination in B-Cell Development. A simplified scheme of B-cell development is presented as a background for discussion of Ig gene recombination. The stages occurring in the bone marrow versus in the periphery (e.g., lymph nodes, spleen) are shown, along with the status of IgH and IgL genes at each stage. A graphic depicting the Ig-related proteins displayed on the surface at each stage is presented; at the bottom, the stage-dependent expression of recombination activating genes and terminal deoxynucleotidyl transferase—both important in V(D)J recombination—is schematically depicted. |
Allelic Exclusion and Regulated V(D)J Recombination
The previous description of B-cell development serves as a background to understand an explanation of allelic exclusion that was first proposed by Alt and colleagues110 and has been supported by subsequent experiments. According to this model the functional rearrangement of an L (or H) chain gene in a particular B cell would inhibit further L (or H) chain gene rearrangement in the same cell. If the inhibition occurred promptly after the first functional rearrangement, then two functional Igs could never be produced in the same cell. An initial nonproductive rearrangement would have no inhibitory effect, so recombination could continue until a functional product resulted or until the cell used up all its germline precursors.
In pro-B cells, the first Ig gene rearrangements join D to JH segments (commonly on both chromosomes), and this is followed by VH to DJH recombination. If the first VH to DJH recombination in a pro-B cell produces a functional VDJ gene, a functional µ H chain will be expressed on the cell surface paired with the SLCs. The expression of this pre-BCR complex has been shown to have two consequences. First, it blocks further H chain recombination by decreasing RAG gene expression111 and by reducing target accessibility, as reflected in decreased VH gene transcription.112 The latter is important for rendering the IgH locus inaccessible during subsequent rearrangement of the Igκ and Igλ loci. If the initial VH to DJH rearrangement is nonfunctional (e.g., out of frame), subsequent VH to DJH recombination occurs on the other allele. If the VDJ recombination product on the
second chromosome is also nonproductive, then the cell has reached a dead end and is eliminated by apoptosis.113
second chromosome is also nonproductive, then the cell has reached a dead end and is eliminated by apoptosis.113
The second consequence of pre-BCR expression is the initiation of Ig L chain recombination. This effect was originally deduced from the rarity of κ-expressing cells without H chain gene rearrangement, suggesting that H chain expression is required for κ recombination. As additional evidence, a functional µ gene introduced into early B-lineage cells can cause RAG gene expression and turn on transcription of unrearranged Vκ genes. These are designated “sterile” transcripts because they cannot encode a κ protein, but they are required for Vκ-Jκ recombination. When this recombination ensues, the possibilities for functional and nonproductive Vκ-Jκ rearrangements resemble those discussed previously for the H chain. Expression of a functional κ chain that can associate with µ to form a surface-expressed IgM molecule results in the downregulation of RAG gene expression and suppression of further κ rearrangements. By this mechanism, functional rearranged VκJ-Cκ transgenes can suppress rearrangement of endogenous κ genes.114
Most B cells show isotypic exclusion (i.e., they express either κ or λ but not both). Furthermore, κ rearrangement seems to occur before λ. Thus in normal and malignant human B-lymphoid cells, κ-expressing cells generally have their λ genes in germline configuration, while in λ-expressing cells, κ genes are either rearranged (rarely) or deleted (most commonly) by recombination signal recombination events discussed previously in this chapter.22 The mechanisms that dictate the order of L chain recombination remain unknown. Plausible models include either the selective suppression of λ recombination until all options on the Igκ locus are exhausted or differences in the timing of the developmental programs controlling κ and λ accessibility.
Regulation of RAG Expression
A complete explanation of RAG gene expression would explain its lymphoid specificity, the two waves of RAG expression (during IgH and IgL rearrangements) and the autoantigen-induced upregulation associated with receptor editing. Although our current knowledge is still incomplete, several cis-regulatory elements that regulate RAG expression have been characterized. Surprisingly, the elements and mechanism for regulating expression during B- and T-cell development are distinct. RAG1 and RAG2 are transcribed toward each other in opposite directions, driven by promoters near the respective transcription start sites. Three B-cell-specific enhancers—designated Erag, D3, and Ep—have been reported, lying about 23 kb, 8 kb, and 1.6 kb, respectively, upstream of RAG2.115,116,117 The B-cell-specific function of these regulatory regions is likely explained by the intersecting specificities of transcription factors that interact with them, including Pax5, E2A, FoxP1, FoxO1, NFATc1, and Ikaros. NFκB, which binds at several locations in the RAG enhancers, and FoxO1 (binding to Erag) were found to be important mediators of the upregulation of RAG expression in cells undergoing receptor editing.118,119 Regulation of RAG2 protein across the cell cycle has been discussed previously in this chapter.
Parameters Affecting Recombinational Accessibility and Transcription
V(D)J recombination is triggered by RAG expression in the development of both B and T cells, yet Ig gene recombination is largely confined to B cells (exception: early T cells typically show D-JH recombination); TCR gene recombination is exclusive to T cells. A widely accepted explanation for this locus specificity is provided by the “accessibility” model.120 This model proposes that only those gene segments programmed for recombination at a given stage of B- and T-cell development are “accessible” to the RAG recombinase. One clue suggesting this model was that susceptibility to recombination and transcription of germline gene elements seem to be tightly correlated.120 For example, many germline VH genes are transcribed at the pre-B cell stage, just at the time when these genes are targets for recombination; these transcripts— designated “sterile” like the Vκ transcripts mentioned previously —are not seen in more mature B cells in which H chain recombination has been terminated. In support of the accessibility model, recombinant RAG proteins incubated with nuclei purified from pro-B cells (which generate sterile transcripts in the IgH locus) were found to cleave DNA at Ig JH RSSs, but not at TCRδ RSSs; conversely, in pro-T nuclei the TCRδ RSS was cleaved, but not an Ig gene RSS.121
One molecular correlate of accessibility is the epigenetic state of DNA in the nuclear chromatin. The minimal repeat unit of chromatin is the nucleosome, which consists of eight core histones (two copies each of H2A, H2B, H3, and H4) with 146 bp DNA wrapped around it. In vitro, RAG proteins are unable to bind to and cut DNA wrapped around nucleosomes,122,123 and hence nucleosomes have to be shifted or removed (a process called chromatin remodeling) to allow access. An alternative but not mutually exclusive approach to gain access is posttranslational modification of the histone tails, which regulates the tightness of DNA-nucleosome contacts. The following section provides an overview of how accessibility of the Ig gene loci for RAG activity is regulated by several distinct but interconnected epigenetic mechanisms. We discuss a few important examples for each mechanism and refer to comprehensive review articles for an in-depth discussion.
Subnuclear Localization
In general, inactive genes tend to be located in the periphery of nuclei, while active genes are recruited to a more central nuclear location.124 It is unclear whether the location per se dictates the chromatin state of a locus or whether the movement is a consequence of a locus being “opened.” Fluorescence in situ hybridization (FISH) with large (˜100 kb) probes specific for Ig loci is routinely used to reveal the position of Ig gene loci and control genes in the nucleus. The IgH and Igκ loci are located at the nuclear periphery in hematopoietic progenitors and pro-T cells, but move to central areas of the nucleus in pro-B cells.125 As only the IgH locus gets rearranged at this stage, the correlation of position with accessibility is not perfect.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


