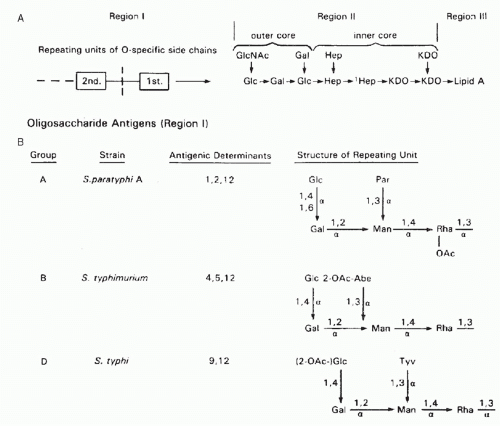
Mapping Epitopes: Conformation versus Sequence
The other component that defines a protein antigenic determinant, besides the amino acid residues involved, is the way these residues are arrayed in three dimensions. As the residues are on the surface of a protein, we can also think of this component as the topography of the antigenic determinant. Sela
42 divided protein antigenic determinants into two categories, sequential and conformational, depending on whether the primary sequence or the three-dimensional conformation appeared to contribute the most to binding. On the other hand, as the antibody-combining site has a preferred topography in the native antibody, it would seem a priori that some conformations of a particular polypeptide sequence would produce a better fit than others and therefore would be energetically favored in binding. Thus, conformation or topography must always play some role in the structure of an antigenic determinant.
Moreover, when one looks at the surface of a protein in a space-filling model, one cannot ascertain the direction of the backbone or the positions of the helices (contrast
Figs. 23.3A and 23.3B).
43,44,45,46,47 It is hard to recognize whether two residues that are side by side on the surface are adjacent on the polypeptide backbone or whether they come from different parts of the sequence and are brought together by the folding of the molecule. If a protein maintains its native conformation when an antibody binds, then it must similarly
be hard for the antibody to discriminate between residues that are covalently connected directly and those connected only through a great deal of intervening polypeptide. Thus, the probability that an antigenic determinant on a native globular protein consists of only a consecutive sequence of amino acids in the primary structure is likely to be rather small. Even if most of the determinant were a continuous sequence, other nearby residues would probably play a role as well. Only if the protein were cleaved into fragments before the antibodies were made would there be any reason to favor connected sequences.
This concept was analyzed and confirmed quantitatively by Barlow et al.,
48 who examined the atoms lying within spheres of different radii from a given surface atom on a protein. As the radius increases, the probability that all the atoms within the sphere will be from the same continuous segment of protein sequence decreases rapidly. Correspondingly, the fraction of surface atoms that would be located at the center of a sphere containing only residues from the same continuous segment falls dramatically as the radius of the sphere increases. For instance, for lysozyme, with a radius of 8 Å, fewer than 10% of the surface residues would lie in such a “continuous patch” of surface. These are primarily in regions that protrude from the surface. With a radius of 10 Å, almost none of the surface residues fall in the center of a continuous patch. Thus, for a contact area of about 20 Å × 25 Å, as found for a lysozyme-antibody complex studied by x-ray crystallography, none of the antigenic sites could be completely continuous segmental sites (see following discussion and
Fig. 23.4). On the other hand, other analyses did not find a correlation of epitope residues with surface accessibility, suggesting that the situation is more complex.
49Antigenic sites consisting of amino acid residues that are widely separated in the primary protein sequence but brought together on the surface of the protein by the way it folds in its native conformation have been called “assembled topographic” sites
50,51 because they are assembled from different parts of the sequence and exist only in the surface topography of the native molecule. By contrast, the sites that consist of only a single continuous segment of protein sequence have been called “segmental” antigenic sites.
50,51In contrast to T-cell recognition of “processed” fragments retaining only primary and secondary structures, the evidence is overwhelming that most antibodies are made against the native conformation when the native protein is used as immunogen. For instance, antibodies to native staphylococcal nuclease were found to have about a 5000-fold higher affinity for the native protein than for the corresponding polypeptide on which they were isolated (by binding to the peptide attached to Sepharose).
52 An even more dramatic example is that demonstrated by Crumpton
53 for antibodies to native myoglobin or to apomyoglobin. Antibodies to native ferric myoglobin produced a brown precipitate with myoglobin but did not bind well to apomyoglobin, which, without the heme, has a slightly altered conformation. On the other hand, antibodies to the apomyoglobin, when mixed with native (brown) myoglobin, produced a white precipitate. These antibodies so strongly favored the conformation of apomyoglobin, from which the heme was excluded, that they trapped those molecules that vibrated toward that conformation and pulled the equilibrium state over to the apo form. One could almost say, figuratively, that the antibodies squeezed the heme out of the myoglobin. Looked at it thermodynamically, it is clear that the conformational preference of the antibody for the apo versus native forms, in terms of free energy, had to be greater than the free energy of binding of the heme to myoglobin. Thus, in general, antibodies are made that are very specific for the conformation of the protein used as immunogen. Other more recent examples also show that antibodies can
enforce structures on disordered or denatured structures in proteins such as HIV-1 Tat
54 or influenza hemagglutinin.
55Synthetic peptides corresponding to segments of the protein antigen sequence can be used to identify the structures bound by antibodies specific for segmental antigenic sites. To identify assembled topographic sites, more complex approaches have been necessary. The earliest was the use of natural variants of the protein antigen with known amino acid substitutions, where such evolutionary variants exist.
50 Thus, substitution of different amino acids in proteins in the native conformation can be examined. The use of this method, which is illustrated later, is limited to studying the function of amino acids that vary among homologous proteins, that is, those that are polymorphic. It may now be extended to other residues by use of site-directed mutagenesis. A second method is to use the antibody that binds to the native protein to protect the antigenic site from modification
56 or proteolytic degradation.
57 A related but less sensitive approach makes use of competition with other antibodies.
58,59,60 A third approach, taking advantage of the capability of producing thousands of peptides on a solid-phase surface for direct binding assays,
61 is to study binding of a monoclonal antibody to every possible combination of six amino acids.
61 If the assembled topographic site can be mimicked by a combination of six amino acids not corresponding to any continuous segment of the protein sequence but structurally resembling a part of the surface, then one can produce a “mimotope” defining the specificity of that antibody.
61 Mimotopes have become widely used and can be combined with mutational analysis to map assembled topographic epitopes.
62 Mimetics have even been made for quaternary structural epitopes.
63 Many mimotope approaches use phase display peptide libraries to map epitopes of monoclonal antibodies.
64,65,66 However, other studies have been less optimistic about the ability to predict assembled topographic or discontinuous epitopes from mimotope binding
67 or random peptide libraries.
68Myoglobin also serves as a good model protein antigen for studying the range of variation of antigenic determinants from those that are more sequential in nature to those that do not even exist without the native conformation of the protein (see
Fig. 23.3). A good example of the first more segmental type of determinant is that consisting of residues 15 to 22 in the amino terminal portion of the molecule. Crumpton and Wilkinson
69 first discovered that the chymotrypsin cleavage fragment consisting of residues 15 to 29 had antigenic activity for antibodies raised to either native or apomyoglobin. Two other groups
44,70 then found that synthetic peptides corresponding to residues 15 to 22 bind antibodies made to native sperm whale myoglobin, even though the synthetic peptides were only seven to eight residues long. Peptides of this length do not spend much time (in solution) in a conformation corresponding to that of the native protein. On the other hand, these synthetic peptides had a several hundred-fold lower affinity for the antibodies than did the native protein. Thus, even if most of the determinant was included in the consecutive sequence 15 to 22, the antibodies were still much more specific for the native conformation of this sequence than for the random conformation peptide. Moreover, there was no evidence to exclude the participation of other residues, nearby on the surface of myoglobin but not in this sequence, in the antigenic determinant.
71,72,73,74*A good example of the importance of secondary structure is the case of the loop peptide (residues 64 to 80) of hen egg white lysozyme.
75 This loop in the protein sequence is created by the disulfide linkage between cysteine residues 64 and 80 and has been shown to be a major antigenic determinant for antibodies to lysozyme.
75 The isolated peptide 60 to 83, containing the loop, binds antibodies with high affinity, but opening of the loop by cleavage of the disulfide bond destroys most of the antigenic activity for antilysozyme antibodies.
75At the other end of the range of conformational requirements are those determinants involving residues far apart in the primary sequences that are brought close together on the surface of the native molecule by its folding in three dimensions, called assembled topographic determinants.
50,51 Of six monoclonal antibodies to sperm whale myoglobin studied by Berzofsky et al.,
43,76 none bound to any of the three cyanogen bromide (CNBr) cleavage fragments of myoglobin that together span the whole sequence of the molecule. Therefore, these monoclonal antibodies (all with affinities between 2 × 10
8 and 2 × 10
9 M
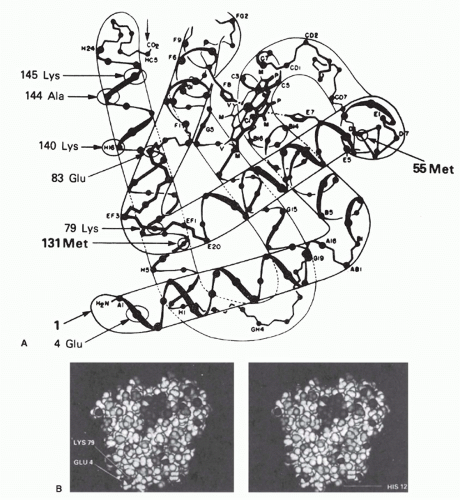
-1) were all highly specific for the native conformation. These were studied by comparing the relative affinities for a series of native myoglobins from different species with known amino acid sequences. This approach allowed the definition of some of the residues involved in binding to three of these antibodies. Two of these three monoclonal antibodies were found to recognize topographic determinants, as defined previously. One recognized a determinant including Glu 4 and Lys 79, which come within about 2 Å of each other to form a salt bridge in the native molecule (see
Fig. 23.3A, B). The other antibody recognized a determinant involving Glu 83, Ala 144, and Lys 145 (see
Fig. 23.3A). Again, these are far apart in the primary sequence but are brought within 12 Å of each other by the folding of the molecule in its native conformation. Similar examples have been reported for monoclonal antibodies to human myoglobin
77 and to lysozyme
37,58 as well as the HIV-1 envelope protein (neutralizing epitopes)
78,79 and the prion protein.
80 Other examples of such conformation-dependent antigenic determinants have been suggested using conventional antisera to such proteins as insulin,
81 hemoglobin,
82 tobacco mosaic virus,
83 and cytochrome c.
84 Moreover, the crystallographic structures of lysozyme-antibody
34,36,37 and neuraminidase-antibody
35 complexes, as well as HIV-1 envelope antibody complexes,
78,79 show clearly that, in both cases, the epitope bound is an assembled topographic site.
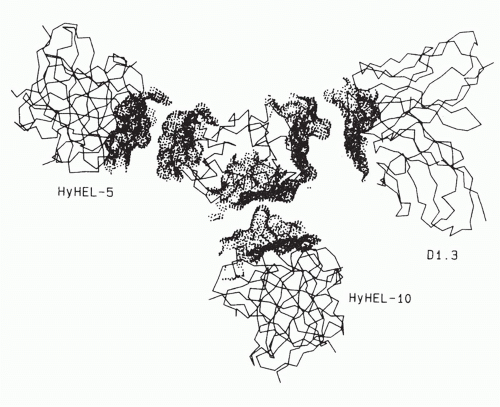
In the case of the three monoclonal antibodies binding to nonoverlapping sites of lysozyme
(Fig. 23.4), it is clear that the footprints of all three antibody-combining sites cover more than one loop of polypeptide chain, and thus, each encompasses an assembled topographic site.
37 This result illustrates the concept that most antibody-combining sites must interact with more than a continuous loop of polypeptide chain and thus must define assembled topographic sites.
48 Another important example is represented by neutralizing antibodies to the HIV envelope protein that similarly bind assembled topographic sites
85,86 (see the end of this section).
How frequent are antibodies specific for topographic determinants compared to those that bind consecutive sequences when conventional antisera are examined? This question was studied by Lando et al.,
87 who passed goat, sheep, and rabbit antisera to sperm whale myoglobin over columns of myoglobin fragments, together spanning the whole sequence. After removal of all antibodies binding to the fragments, 30% to 40% of the antibodies remained that still bound to the native myoglobin molecule with high affinity but did not bind to any of the fragments in solution by radioimmunoassay. Thus, in four of four antimyoglobin sera tested, 60% to 70% of the antibodies could bind peptides, and 30% to 40% could bind only native-conformation intact protein.
On the basis of studies such as these, it has been suggested that much of the surface of a protein molecule may be antigenic,
50,88 but that the surface can be divided up into antigenic domains.
43,73,74,77 Each of these domains consists of many overlapping determinants recognized by different antibodies.
An additional interesting point is that in three published crystal structures of protein antigen-antibody complexes, the contact surfaces were broad, with local complementary pairs of concave and convex regions in both directions.
34,35,36,37 Thus, the concept of an antigen binding in the groove or pocket of an antibody may be oversimplified, and antibodies may sometimes bind by extending into pockets on an antigen.
Further information on the subjects discussed in this section is available in the reviews by Sela,
42 Crumpton,
53 Reichlin,
89 Kabat,
90 Benjamin et al.,
50 Berzofsky,
51 Getzoff et al.,
41 and Davis and Padlan.
37
Antipeptide Antibodies that Bind to Native Proteins at a Specific Site
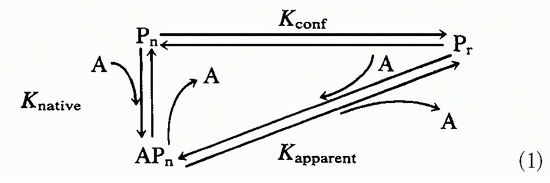
In light of the conformational differences between native proteins and peptides and the observed K
conf effects shown by antibodies to native proteins when tested on the corresponding peptides, it was somewhat surprising to find that antibodies to synthetic peptides show extensive cross-reactions with native proteins.
94,95 These two types of cross-reactions can be thought of as working in opposite directions: The binding of antiprotein antibodies to the peptide is inefficient, whereas the binding of antipeptide antibodies to the protein is quite efficient and commonly observed. This finding is quite useful, as automated solid-phase peptide synthesis has become readily available. This has been particularly useful in three areas: exploitation of protein sequences deduced by recombinant deoxyribonucleic acid (DNA) methods, preparation of site specific antibodies, and the attempt to focus the immune response on a single protein site that is biologically important but may not be particularly immunogenic. This section focuses on the explanation of the cross-reaction, uses of the cross-reaction, and the potential limitations regarding immunogenicity.
The basic assumption is that antibodies raised against peptides in an unfolded structure will bind the corresponding site on proteins folded into the native structure.
95 This is not immediately obvious, as antibody binding to antigen is the direct result of the antigen fitting into the binding site. Affinity is the direct consequence of “goodness of fit” between antibody and antigen, whereas antibody specificity is due to the inability of other antigens to occupy the same site. How then can the antipeptide antibodies overcome the effect of K
conf and still bind native proteins with good affinity and specificity? The whole process depends on the antibodybinding site forming a three-dimensional space and the antigen filling it in an energetically favorable way.
Because the peptides are randomly folded, they rarely occupy the native conformation, so they are not likely to elicit antibodies against a conformation they do not maintain. If the antibodies are specific for a denatured structure, then, like the myoglobin molecules that were denatured to apomyoglobin by antibody binding,
53 the cross-reaction may depend on the native protein’s ability to assume different conformational states. If the native protein is quite rigid, then the possibility of it assuming a random conformation is quite small; if it is a flexible three-dimensional spring, then local unfolding and refolding may occur all the time. Local unfolding of protein segments may permit the immunologic cross-reaction with antipeptide antibodies, as a flexible segment could assume many of the same conformations as the randomly folded peptide.
95 On the other hand, peptides with more stable conformations may be more likely to elicit antibodies that bind both the peptide and the native protein.
96 To this end, scaffolding has been used to maintain the conformation of peptides or protein fragments to be used as immunogens/vaccines, such as for respiratory syncytial virus
97 or HIV epitopes.
98,99In contrast, the ability of proteins to crystallize (a feature that allows the study of their structure by x-ray crystallography) has long been taken as evidence of protein rigidity.
100 In addition, the existence of discrete functional states of allosteric enzymes
101 provides additional evidence of stable structural states of a protein. Finally, the fact that antibodies can distinguish native from denatured forms of intact proteins is well known for proteins such as myoglobin.
53However, protein crystals are a somewhat artificial situation, as the formation of the crystal lattice imposes order on the components, each of which occupies a local energy minimum at the expense of considerable loss of randomness (entropy). Thus, the crystal structure may have artificial rigidity that exceeds the actual rigidity of protein molecules in solution. On the contrary, we may attribute some of the considerable difficulty in crystallizing proteins to disorder within the native conformation. Second, allosterism may be explained by two distinct conformations that are discrete without being particularly rigid. Finally, the ability to generate antiprotein antibodies that are conformation specific does not rule out the existence of antipeptide antibodies that are not. All antibodies are probably specific for some conformation of the antigen, but this need not be the crystallographic native conformation in order to achieve a significant affinity for those proteins or protein segments that have a “loose” native conformation.
Antipeptide antibodies have proved to be very powerful reagents when combined with recombinant DNA methods of gene sequencing.
95,102 From the DNA sequence, the protein sequence is predicted. A synthetic peptide is constructed, coupled to a suitable carrier molecule, and used to immunize animals. The resulting polyclonal antibodies can be detected with a peptide-coated enzyme-linked immunosorbent assay plate (see
Chapter 7). They are used to immunoprecipitate the native protein from a
35S-labeled cell lysate and thus confirm expression of the gene product in these cells. The antipeptide antibodies can also be used to isolate the previously unidentified gene product of a new gene. The site-specific antibodies are also useful in detecting posttranslational processing, as they bind all precursors and products that contain the site. In addition, because the antibodies bind only to the site corresponding to the peptide, they are useful in probing structure-function relationships. They can be used to block the binding of a substrate to an enzyme or the binding of a virus to its cellular receptor.
Immunogenicity of Proteins and Peptides
Up to this point, we have considered the ability of antibodies to react with proteins or peptides as antigens. However, immunogenicity refers to the ability of these compounds to elicit antibodies following immunization. Several factors limit the immunogenicity of different regions of proteins, and these have been divided into those that are intrinsic to protein structure itself versus those extrinsic to the antigen that are related to the responder and vary from one animal or species to another.
51 In addition, we consider the special case of peptide immunogenicity, as it applies to vaccine development. The features of protein structure that have been suggested to explain the results include surface accessibility
of the site, hydrophilicity, flexibility, and proximity to a site recognized by helper T cells.
When the x-ray crystallographic structure and antigenic structure are known for the same protein, it is not surprising to find that a series of monoclonal antibodies binding to a molecule such as influenza neuraminidase choose an overlapping pattern of sites at the exposed head of the protein.
103The stalk of neuraminidase was not immunogenic apparently because it was almost entirely covered by carbohydrate. Beyond such things as carbohydrate, which may sterically interfere with antibody binding to protein, accessibility on the surface is clearly a sine qua non for an antigenic determinant to be bound by an antibody specific for the native conformation, without any requirement for unfolding of the structure.
51 Several measures of such accessibility have been suggested. All these require knowledge of the x-ray crystallographic three-dimensional structure. Some have measured accessibility to solvent by rolling a sphere with the radius of a water molecule over the surface of a protein.
104,105 Others have suggested that accessibility to water is not the best measure of accessibility to antibody and have demonstrated a better correlation by rolling a sphere with the radius of an antibody-combining domain.
106 Another approach to predicting antigenic sites on the basis of accessibility is to examine the degree of protrusion from the surface of the protein.
107 This was done by modeling the body of the protein as an ellipsoid and examining which amino acid residues remain outside ellipsoids of increasing dimensions. The most protruding residues were found to be part of antigenic sites bound by antibodies, but usually, these sites had been identified by using short synthetic peptides and so were segmental in nature. As noted previously, for an antigenic site to be contained completely within a single continuous segment of protein sequence, the site is likely to have to protrude from the surface, as otherwise residues from other parts of the sequence would fall within the area contacting the antibody.
48 However, inability of such surface or protrusion information to predict antigenic sites has also been encountered in some studies.
49Because the three-dimensional structure of most proteins is not known, other ways of predicting surface exposure have been proposed for the vast majority of antigens. For example, hydrophilic sites tend to be found on the water-exposed surface of proteins. Thus, hydrophilicity has been proposed as a second indication of immunogenicity.
108,109,110 This model has been used to analyze 12 proteins with known antigenic sites: The most hydrophilic site of each protein was indeed one of the antigenic sites. However, among the limitations are the facts that a significant fraction of surface residues can be nonpolar,
104,105 and that several important examples of hydrophobic and aromatic amino acids involved in the antigenic sites are known.
42,83,111,112 Specificity of antibody binding likely depends on the complementarity of surfaces for hydrogen bonding and polar bonding as well as van der Waals contacts, whereas hydrophobic interactions and the exclusion of water from the interacting surfaces of proteins may contribute a large but nonspecific component to the energy of binding.
113 Another study suggested that amino acid pairs were better predictors of epitopes.
114A third factor suggested to play a role in immunogenicity of protein epitopes is mobility. Measurement of mobility in the native protein is largely dependent on the availability of a high-resolution crystal structure, so its applicability is limited to only a small subset of proteins. Furthermore, it has been studied only for antibodies specific for segmental antigenic sites; therefore, it may not apply to the large fraction of antibodies to assembled topographic sites. Studies of mobility have taken two directions. The case of antipeptide antibodies has already been discussed, in which antibodies made to peptides corresponding to more mobile segments of the native protein were more likely to bind to the native protein.
95,115 This is not considered just a consequence of the fact that more mobile segments are likely to be those on the surface and therefore more exposed because in the case of myohemerythrin (which was used as a model), two regions of the native protein that were equally exposed but less mobile did not bind nearly as well to the corresponding antipeptide antibodies.
116 However, as is clear from the previous discussion, this result applies to antibodies made against short peptides and therefore is not directly relevant to immunogenicity of parts of the native protein. Rather, it concerns the cross-reactivity of antipeptide antibodies with the native protein and therefore is of considerable practical importance for the purposes outlined in the section on antipeptide antibodies.
Studies in the other direction—that is, of antibodies raised against native proteins—would be by definition more relevant to the question of immunogenicity of parts of the native protein. Westhof et al.
117 used a series of hexapeptides to determine the specificity of antibodies raised against native tobacco mosaic virus protein and found that six of the seven peptides that bound antibodies to native protein corresponded to peaks of high mobility in the native protein. The correlation was better than could be accounted for just by accessibility because three peptides that corresponded to exposed regions of only average mobility did not bind antibodies to the native protein. However, when longer peptides—on the order of 20 amino acid residues—were used as probes, it was found that antibodies were present in the same antisera that bound to less mobile regions of the protein.
118 They simply had not been detected with the short hexapeptides with less conformational stability. Thus, it was not that the more mobile regions were necessarily more immunogenic but rather that antibodies to these were more easily detected with short peptides as probes. A similar good correlation of antigenic sites with mobile regions of the native protein in the case of myoglobin
117 may also be attributed to the fact that seven of the nine sites were defined with short peptides of six to eight residues.
71 Again, this result becomes a statement about cross-reactivity between peptides and native protein rather than about the immunogenicity of the native protein. For reviews, see Van Regenmortel
119 and Getzoff et al.
41To address the role of mobility in immunogenicity, an attempt was made to quantitate the relative fraction of antibodies specific for different sites on the antigen myohemerythrin.
120 The premise was that, although the entire surface of the protein may be immunogenic, certain regions may elicit significantly more antibodies than others and therefore may
be considered immunodominant or at least more immunogenic. Because this study was done with short synthetic peptides from 6 to 14 residues long based on the protein sequence, it was limited to the subset of antibodies specific for segmental antigenic sites. Among these, it was clear that the most immunogenic sites were in regions of the surface that were most mobile, convex in shape, and often of negative electrostatic potential. Other more recent studies corroborate the greater immunogenicity of more flexible segments of protein structures.
121 The role of these parameters has been reviewed.
41These results have important practical and theoretical implications. First, to use peptides to fractionate antiprotein antisera by affinity chromatography, peptides corresponding to more mobile segments of the native protein should be chosen when possible. If the crystal structure is not known, it may be possible to use peptides from amino or carboxyl termini or from exon-intron boundaries, as these are more likely to be mobile.
115 Second, these results may explain how a large but finite repertoire of antibody-producing B cells can respond to any antigen in nature or even artificial antigens never encountered in nature. Protein segments that are more flexible may be able to bind by induced fit in an antibody-combining site that is not perfectly complementary to the average native structure.
41,51 Indeed, evidence from the crystal structure of antigen-antibody complexes
122,123,124 suggests that mobility in the antibody-combining site as well as in the antigen may allow both reactants to adopt more complementary conformations on binding to each other, that is, a two-way induced fit. A nice example comes from the study of antibodies to myohemerythrin,
123 in which the data suggested that initial binding of exposed side chains of the antigen to the antibody promoted local displacements that allowed exposure and binding of other, previously buried residues that served as contact residues. The only way this could occur would be for such residues to become exposed during the course of an induced fit conformational change in the antigen.
41,123 In a second very clear example of induced fit, the contribution of antibody mobility to peptide binding was demonstrated for a monoclonal antibody to peptide 75 to 110 of influenza hemagglutinin, which was crystallized with or without peptide in the binding site and analyzed by x-ray crystallography for evidence of an induced fit.
124 Despite flexibility of the peptide, the antibody-binding site probably could not accommodate the peptide without a conformational change in the third complementarity determining region of the heavy chain, in which an asparagine residue of the antibody was rotated out of the way to allow a tyrosine residue of the peptide to fit in the binding pocket of the antibody.
124Regarding host-limited factors, immunogenicity is certainly limited by self-tolerance. Thus, the repertoire of potential antigenic sites on mammalian protein antigens such as myoglobin or cytochrome c can be thought of as greatly simplified by the sharing of numerous amino acids with the endogenous host protein. For mouse, guanaco, or horse cytochrome c injected into rabbits, each of the differences between the immunogen and rabbit cytochrome c is seen as an immunogenic site on a background of immunologically silent residues.
50,84,125 In another example, rabbit and dog antibodies to beef myoglobin bound almost equally well to beef or sheep myoglobin.
126 However, sheep antibodies bound beef but not sheep myoglobin, even though these two myoglobins differ by just six amino acids. Thus, the sheep immune system was able to screen out those clones that would be autoreactive with sheep myoglobin.
Ir genes of the host also play an important role in regulating the ability of an individual to make antibodies to a specific antigen.
127 These antigen-specific immune response genes are among the major histocompatibility complex (MHC) genes that code for transplantation antigens. Structural mutations, gene transfer experiments, and biochemical studies
127 all indicate that Ir genes are actually the structural genes for MHC antigens. The mechanism of action of the MHC antigens works through their effect on helper T cells (described later in this chapter). There appear to be constraints on which B and T cells of a given specificity can help,
128,129 a process called T-B reciprocity.
130 Thus, if Ir genes control helper T-cell specificity, they will in turn limit which B cells are activated and which antibodies are made.
The immunogenicity of peptide antigens is also limited by intrinsic and extrinsic factors. With less structure to go on, each small peptide must presumably contain some non-self-structural feature in order to overcome self-tolerance. In addition, the same peptide must contain antigenic sites that can be recognized by helper T cells as well as by B cells. When no T-cell site is present, three approaches may be helpful: graft on a T-cell site, couple the peptide to a carrier protein, or overcome T-cell nonresponsiveness to the available structure with various immunologic agents, such as interleukin 2.
An example of a biologically relevant but poorly immunogenic peptide is the asparagine-alanine-asparagine-proline (NANP) repeat unit of the circumsporozoite (CS) protein of malaria sporozoites. A monoclonal antibody to the repeat unit of the CS protein can protect against murine malaria.
131 Thus, it would be desirable to make a malaria vaccine of the repeat unit of
Plasmodium falciparum (NANP)
n. However, only mice of one MHC type (H-2
b) of all mouse strains tested were able to respond to (NANP)
n.
132,133 One approach to overcome this limitation is to couple (NANP)
n to a site recognizable by T cells, perhaps a carrier protein such as tetanus toxoid.
134 In human trials, this conjugate was weakly immunogenic and only partially protective. Moreover, as helper T cells produced by this approach are specific for the unrelated carrier, a secondary or memory response would not be expected to be elicited by the pathogen itself.
Another choice might be to identify a T-cell site on the CS protein itself and couple the two synthetic peptides together to make one complete immunogen. The result with one such site, called Th2R, was to increase the range of responding mouse MHC types by one, to include H-2
k as well as H-2
b.
135 This approach has the potential advantage of inducing a state of immunity that could be boosted by natural exposure to the sporozoite antigen. As CS-specific T and B cells are both elicited by the vaccine, natural exposure to the antigen could help maintain the level of immunity during the entire period of exposure.
Another strategy to improve the immunogenicity of peptide vaccines is to stimulate the T- and B-cell responses artificially by adding interleukin 2 to the vaccine. Results with myoglobin indicate that genetic nonresponsiveness can be overcome by appropriate doses of interleukin 2.
136 The same effect was found for peptides derived from malaria proteins.
137,137aOne of the most important possible uses of peptide antigens is as synthetic vaccines. However, even though it is possible to elicit with synthetic peptides anti-influenza antibodies to nearly every part of the influenza hemagglutinin,
94 antibodies that neutralize viral infectivity have not been elicited by immunization with synthetic peptides. This may reflect the fact that antibody binding by itself often does not result in virus inactivation. Viral inactivation occurs only when antibody interferes with one of the steps in the life cycle of the virus, including binding to its cell surface receptor, internalization, and virus uncoating within the cell. Apparently, antibodies can bind to most of the exposed surface of the virus without affecting these functions. Only those antibodies that bind to certain “neutralizing” sites can inactivate the virus. In addition, as in the case of the VP1 coat protein of poliovirus, certain neutralizing sites are found only on the native protein and not on the heat-denatured protein.
138 Thus, not only the site but also the conformation that is bound by the antibodies may be important for the antibody to inactivate the virus. These sites may often be assembled topographic sites not mimicked by peptide segments of the sequence. Perhaps binding of an antibody to such an assembled site can alter the relative positions of the component subsites so as to induce an allosteric neutralizing effect. Alternatively, antibodies to such an assembled site may prevent a conformational change necessary for activity of the viral protein.
One method of mapping neutralizing sites is based on the use of neutralizing monoclonal antibodies. The virus is grown in the presence of neutralizing concentrations of the monoclonal antibody, and virus mutants are selected for the ability to overcome antibody inhibition. These are sequenced, revealing the mutation that permits “escape” by altering the antigenic site for that antibody. This method has been used to map the neutralizing sites of influenza hemagglutinin
139 as well as poliovirus capsid protein VP1.
140 The influenza escaping mutations are clustered to form an assembled topographic site, with mutations distant from each other in the primary sequence of hemagglutinin but brought together by the three-dimensional folding of the native protein. At first, it was thought that neutralization was the result of steric hindrance of the hemagglutinin-binding site for the cell surface receptor of the virus.
141 However, similar work with poliovirus reveals that neutralizing antibodies that bind to assembled topographic sites may inactivate the virus at less than stoichiometric amounts, when at least half of the sites are unbound by antibody.
142 The neutralizing antibodies all cause a conformational change in the virus, which is reflected in a change in the isoelectric point of the particles from pH 7 to pH 4.
140,143 Antibodies that bind without neutralizing do not cause this shift. Thus, an alternative explanation for the mechanism of antibody-mediated neutralization is the triggering of the virus to self-destruct. Perhaps the reason that neutralizing sites are clustered near receptor-binding sites is that occupation of such sites by antibody mimics events normally caused by binding to the cellular receptor, causing the virus to prematurely trigger its cell entry mechanisms. However, in order to transmit a physiologic signal, the antibody may need to bind viral capsid proteins in the native conformation (especially assembled topographic sites), which antipeptide antibodies may fail to do. Antibodies of this specificity are similar to the viral receptors on the cell surface, some of which have been cloned and expressed without their transmembrane sequences as soluble proteins. The soluble recombinant receptors for poliovirus
144 and HIV-1
145,146,147 exhibit high-affinity binding to the virus and potent neutralizing activity in vitro. The HIV-1 receptor, cluster of differentiation (CD)4, has been combined with the human Ig heavy chain in a hybrid protein CD4-Ig,
148 which spontaneously assembles into dimers and resembles a monoclonal antibody, in which the binding site is the same as the receptor-binding site for HIV-1. In these recombinant constructs, high-affinity binding depends on the native conformation of the viral envelope glycoprotein gp120. Binding of CD4 to gp120 elicits a conformational change exposing a CD4-induced epitope, and fusions of CD4 domains to gp120 can be used as vaccines to elicit such antibodies.
149For HIV-1, two types of neutralizing antibodies have been identified. The first type binds a continuous or segmental determinant, such as the “V3 loop” sequence between amino acids 296 and 331 of gp120.
150,151,152 Antipeptide antibodies against this site can neutralize the virus.
150 However, because this site is located in a highly variable region of the envelope, these antibodies tend to neutralize a narrow range of viral variants with nearly the same sequence as the immunogen. Even for this highly variable site, more broadly neutralizing antibodies can be obtained that recognize conserved conformations.
153,154,155 The second type of neutralizing antibody binds conserved sites on the native structure of gp120, allowing them to neutralize a broad spectrum of HIV-1 isolates. These antibodies are commonly found in the sera of infected patients,
156 and a panel of neutralizing monoclonals derived from these subjects has been analyzed.
These monoclonals can be divided into three types. One group, possibly the most common ones in human polyclonal sera, bind at or near the CD4 receptor-binding site of gp120.
79,157,158,159,160,161 A second type of monoclonal, called 2G12, binds a conformational site on gp120 that also depends on glycosylation, but has no direct effect on CD4 binding.
162 A third type, quite rare in human sera, is represented by monoclonal antibody 2F5
163 and binds a conserved site on the transmembrane protein gp41. Although this site is contained on a linear peptide ELDKWA, antibodies such as 2F5 cannot be elicited by immunizing with the peptide, again suggesting the conformational aspect of this site.
164,165 Indeed, the binding of antibodies to this membrane-proximal site has even been found to involve interaction of the antibody with the lipid membrane.
166 One might view this intriguing case as an example of a discontinuous or assembled topographic site created by the proximity of residues of the protein with structures in the lipid membrane, thus, spanning more than just different parts of the antigenic protein.
These monoclonals neutralize fresh isolates, as well as laboratory-adapted strains, and they neutralize viruses tropic for T cells or macrophages,
167 regardless of the use of CXCR4 or CCR5 as second receptor. These monoclonals, which target different sites, act synergistically. A cocktail combining all three types of monoclonals can protect monkeys against iv challenge or vaginal challenge with a simian immunodeficiency virus/HIV hybrid virus, indicating the potential for antibodies alone to prevent HIV infection.
168,169 Because each of the three conserved neutralizing determinants depends on the native conformation of the protein,
170 a prospective gp120 vaccine (or gp160 vaccine) would need to be in the native conformation to be able to elicit these antibodies.



 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

