22 Immunobiology and Immune Therapy
Glioblastoma multiforme (GBM) is the most common and most aggressive primary malignant brain tumor. Despite numerous advances in therapy, including image-guided surgical resection, high-dose external beam radiotherapy, antiangiogenic treatments, and chemotherapy, patients with GBM generally live less than 15 months from the time of diagnosis.1 Moreover, these treatments are often nonspecific and result in incapacitating damage to surrounding normal brain and systemic tissues. A promising alternative is the use of the immune system, which has the theoretical capacity to eliminate neoplastic cells while leaving healthy cells intact. Although first proposed over a century ago, the concept of antitumor immunotherapy has historically struggled to successfully translate effective treatments for patients with cancer. This changed dramatically in 2010 after pivotal approvals by the Food and Drug Administration for immune-based cancer treatments, sipuleucel-T and ipilimumab,2 in the treatment of hormone-refractory prostate cancer and metastatic melanoma, respectively. Moreover, in human brain tumors, several novel tumor-specific antigens have been identified,3–7 lending further credence to the advancement of immunotherapy as a standard-of-care treatment for GBM. A number of approaches are currently in development toward this end, and a broad understanding of their applications, particularly in the context of intracerebral tumors, represents an emerging priority.
Pearl
• After several decades, the long-standing goal of activating the immune system for therapeutic benefit in cancer has recently been proven in clinical trials to yield significant survival benefits in patients with malignant disease.
 Central Nervous System Immune Privilege
Central Nervous System Immune Privilege
One of the most prominent challenges that has hampered the successful translation of immunotherapies for intracerebral tumors is that of central nervous system (CNS) immune privilege. The notion of limited immune surveillance in the CNS was suggested by several studies in the early 20th century, and was perhaps most clearly demonstrated in 1948 by Sir Peter Medawar, who showed that allogeneic tissue grafts were not rejected following transplantation into the brains of experimental animals. Over the decades that followed, additional work would suggest that these findings were likely due to several characteristics now commonly attributed to the CNS—namely, the presence of a specialized blood–brain barrier (BBB), the lack of conventional lymphatics or resident antigen-presenting cells, and generally low expression of human leukocyte antigens (HLAs) in the brain.
Although the CNS certainly exhibits aspects of immune privilege to some degree, a growing body of literature suggests that CNS isolation from the immune system is not as complete as was once believed. It is now known, for example, that CNS antigen egress occurs along cerebrospinal fluid compartments toward cervical lymph nodes, and that several resident glial cells have the capacity to mediate HLA-restricted antigen presentation as surrogate antigen-presenting cells in the brain. Moreover, despite the BBB, both antibodies8 and immune cells have been shown to penetrate and contribute to routine immune surveillance throughout the CNS. Intracerebral immune access is perhaps most convincingly demonstrated by the presence of tumor-infiltrating lymphocytes in patients with GBM and the positive association between intratumoral T cells and clinical outcome.9 Importantly, deterioration of the BBB has also been well documented in the setting of glioma, which may contribute to the freer penetration of immune cells, especially in areas of frank neural inflammation.
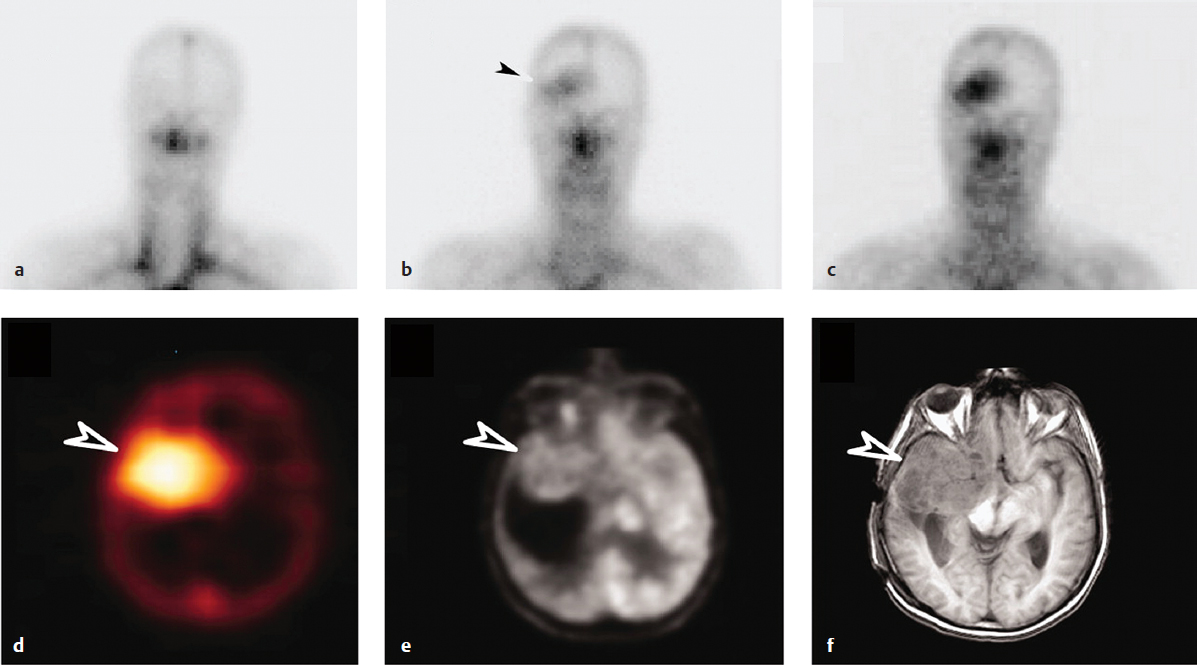
As alluded to above, large molecules such as antibodies have also been shown to localize to intracerebral tumors.8 In addition to tumor-mediated BBB disruption, one leading explanation for this phenomenon is the emerging “antigen-sink hypothesis,” which states that antibodies and other macromolecules can accumulate beyond the BBB in the proximity of their cognate antigen, if and only if this cognate antigen is not also expressed in other tissues outside the CNS. Because basal levels of nonspecific peripherally circulating antibodies can be detected in the CNS under normal conditions (between 0.1% and 1% of that found in serum), it is thought that, in the absence of cross-reactivity with systemic antigens, small amounts of highly tumor-specific proteins will penetrate the CNS through passive diffusion, and be retained there over time to reach therapeutically relevant quantities. This theory has been corroborated by studies in which systemic infusion with a monoclonal antibody against a tumor-specific mutant of the epidermal growth factor receptor, EGFRvIII, resulted in elevated levels of intracerebral uptake in patients with GBM compared to antibodies possessing cross-reactivity against systemic antigen (Fig. 22.1).10 Moreover, vaccine-induced humoral immunity to EGFRvIII has been shown to lead to complete elimination of EGFRvIII expression in recurrent tumors of patients with GBM,11 providing further evidence of the capacity of antibody responses in the periphery to mediate therapeutic effects beyond the BBB.
Pearl
• Despite conventional notions of CNS immune privilege, growing evidence suggests that both immune cells and antibodies have the capacity to penetrate and treat tumors in the brain.
 Glioma-Induced Immune Suppression
Glioma-Induced Immune Suppression
Gliomas are also known to potentiate a hostile environment that can antagonize the development of efficacious antitumor immune responses. Immune suppression in patients with GBM is manifested by several metrics including low peripheral lymphocyte counts, depressed skin reactivity to recall antigens, and impaired T-cell function with a counterproductive Th2 cytokine skew. A number of pathways have been shown to contribute to the development of this pro-tumorigenic milieu. Like other cancers, gliomas secrete immunosuppressive cytokines including prostaglandin E2 (PGE2), transforming growth factor-β (TGF)-β, and interleukin-10 (IL-10), leading to potentiation of immune resistance through downregulation of major histocompatibility complex (MHC) expression and suppression of both normal T-cell proliferation and dendritic cell (DC) maturation.
The catabolic enzyme indoleamine 2,3-dioxygenase (IDO) has also been shown to possess immunosuppressive and tolerogenic effects in cancer, in part through the depletion of essential tryptophan stores.12 In addition to modulation of soluble factors, specific subsets of immune regulatory cells have been cited as central mediators of tumor-associated immune suppression. Among these are tumor-associated myeloid-derived suppressor cells (MDSCs) and macrophages, as well as a subset of T cells known as regulatory T cells (Tregs), which perhaps have been most frequently implicated. Not only have Tregs been shown to be present in increased proportions both the peripheral blood and tumors of patients with GBM,13,14 but also their presence has been correlated with the overall malignant behavior of these tumors.15 Tregs are known to suppress effector T-cell responses, specifically by restricting IL-2 production and inducing T-cell anergy. Importantly, Treg depletion has been positively associated with enhanced immunity in patients with GBM,16 and in vivo impairment in tumor-bearing mice has been shown to prolong survival.
• Successful translation of immunotherapy for GBM will have to carefully account for the complex interplay among the several immune suppressive mechanisms, including tumor-secreted factors (e.g., TGF-β), cell-surface proteins (e.g., B7-H1), immunoregulatory leukocytes (e.g., Tregs, MDSCs), and cell-signaling pathways (e.g., STAT3), in these patients.
Gliomas not only possess low levels of surface MHC, but also are known to express co-stimulatory inhibitory molecules that may lead to direct inhibition of immune responses.17 These interactions may be manipulated by GBM cells to unfavorably alter signaling through immune checkpoints such as programmed cell death (PD)-1 and cytotoxic T-lymphocyte antigen (CTLA)-4. Importantly, CTLA-4 blockade has been demonstrated as an effective therapy in murine models of GBM18 as well as in patients with melanoma brain metastases.19 Other areas of investigation have underscored the role of signal transducer and activator of transcription-3 (STAT3) in regulating inflammatory responses. Interestingly, the STAT3 pathway in both immune cells and tumor cells has been shown to potentiate immune suppression, leading to generalized impairment of effector responses. As such, therapies designed to modulate STAT3 as well as co-stimulatory signaling18 to enhance antitumor immune responses are among many promising therapies for GBM under current investigation.
 Glioma-Associated Tumor Antigens
Glioma-Associated Tumor Antigens
It is likely that as immunotherapies become more potent, the risk of adverse immune side effects may increase. This is especially true for immunotherapeutic platforms designed to target glioma-associated antigens that are shared with normal, healthy brain tissue; active immunization against these antigens could escalate the risk of uncontrolled CNS auto-immunity, similar to what has been observed in preclinical models of experimental allergic encephalomyelitis (EAE). Importantly, following vaccination with tissue derived from human gliomas, lethal EAE or EAE-like toxicity has been documented both in nonhuman primates as well as in human studies of immunotherapy for brain tumors. These results are to some degree expected, given that gliomas are known to express both normal adult and fetal brain antigens, and because several preclinical protocols are already known to produce EAE upon immunization with CNS tissue.
Table 22.1 Conserved Tumor-Specific Mutations in Brain Tumors
| Protein | Mutation | Function |
| EGFR10 | EGFRvIII | Constitutively activated form of EGFR, promotes cell proliferation, inhibits apoptosis, resistance to radiation and chemotherapy |
| BRAF10,49 | f-BRAF | Promotes cell proliferation, differentiation, and survival |
| H3.35 | K27M, G34R/V | Implicated in chromatin remodeling and telomere architecture |
| IDH14 | R132H | Central metabolism, oxidative decarboxylation of isocitrate to 2-oxoglutarate |
The risk of autoimmune toxicity represents an ongoing concern for tumor vaccines that are not carefully selected for tumor specificity. However, this issue can be avoided to some extent by redirecting immune responses against antigens that are expressed solely in gliomas and completely absent from all normal tissues. To date, a number of such antigens have been characterized (Table 22.1). Among these are the tumorspecific mutant protein, EGFRvIII,10 as well as frequent and homogeneous mutations associated with isocitrate dehydrogenase 1 (IDH1).4 More recently, conserved tumor-specific mutations in H3.35 and BRAF3,20 have also been isolated as potential targets in pediatric GBM and pilocytic astrocytomas, respectively. Also notable are viral antigens unique to human cytomegalovirus (CMV), which have been shown to be expressed in a high percentage of GBM tumors but not surrounding normal brain.21 Interestingly, although the presence of CMV proteins (e.g., IE1, pp65, gB) in gliomas has been confirmed by several independent groups, there have been occasional reports of failure to detect CMV antigen as well, likely owing to methodological differences and the low levels of CMV antigen expression associated with these tumors.22
In addition, several nonrecurrent tumor-specific mutations have been identified, particularly in pediatric GBM; these include sporadic variants in ATRX/DAXX,5,6 TP53,5 NF1,7 and PDGFRA.7 Almost certainly, advances in high-throughput genomic screening technologies will continue to reveal additional antigens that may serve as tumor-specific targets, the discovery of which would represent a potential boon for the further development of safe, effective immunotherapies for glioma.
Pitfall
• Although immunotherapeutic targeting of single tumorspecific mutations holds promise, the inherent heterogeneity of malignant gliomas may require implementation of broader approaches, which in turn must be balanced against the risk of autoimmunity against normal tissues.
 Glioblastoma Vaccines and Immunotherapeutic Approaches
Glioblastoma Vaccines and Immunotherapeutic Approaches
The overall goal of tumor immunotherapy is to develop or enhance immune responses against established disease, even in cases where natural immune surveillance has either failed or been rendered less effective by mechanisms of tumor-associated immune evasion. To date, a wide range of immune-based strategies have been developed toward this end. Among these are passive infusion with antibodies as well as adoptive transfer of tumor-reactive lymphocytes, cytokine-based stimulatory treatments, and an array of active immunization methods in the form of vaccines administered to elicit host immunity against tumor cells. The following subsections introduce the basic principles of several of these modalities, with a focus on major advances that have emerged from representative clinical trials.
Active Immunotherapy
Active immunotherapies seek to achieve priming of host immunity and immunologic memory against tumors through the use of vaccines (Fig. 22.2). Over the years, approaches to establishing active immunotherapy against brain tumors have varied widely, and in some cases have led to successful advancement toward phase III randomized studies.
Autologous Tumor Cell Vaccines
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


