Virus Classification
According to the International Committee of Taxometry of Viruses, all viruses are classified into order, family, subfamily, genus, and species. Names of serotypes, genotypes, strains, variants, or isolates of virus species or artificial viruses are not ruled by the International Committee of Taxometry of Viruses. A species is defined as a polythetic class of viruses that constitutes a replicating lineage and occupies a particular ecologic niche. A genus defines a group of species that share common characteristics, while subfamilies and families define a group of genera with common characteristics. An order, in turn, defines families with shared characteristics. Currently, 6 orders, 87 families, 19 subfamilies, 348 genera, and 2288 species of virus have been defined.
5 The Baltimore classification differs and divides viruses according to their genome or their mode of replication. Viruses contain either a deoxyribonucleic acid (DNA) or ribonucleic acid (RNA) genome, which can be single stranded (ss) or double stranded (ds). SsRNA viruses carry either a negative- or positive-stranded RNA. During their lifecycle, some DNA and RNA viruses undergo an intermediate step in which the RNA genome is converted by reverse transcription (RT) into DNA or vice versa their DNA genome into an RNA genome. Accordingly, viruses are classified into dsDNA viruses (eg, adeno or poxviruses), ssDNA viruses (eg, parvoviruses), dsRNA viruses (eg, reovirus), positive-sense ssRNA viruses (eg, picornaviruses), negativesense ssRNA viruses (eg, rhabdoviruses), positive-sense ssRNA-RT viruses (eg, retroviruses), or dsDNA-RT viruses (eg, hepadnaviruses). Other classifications are based on host range (Holmes classification) or structural characteristics (Lworff-Horne-Tournier [LHT] system).
The viral genome can be linear (eg, poxvirus), circular (eg, papillomaviruses), or segmented (eg, influenza virus). Viruses are further classified into enveloped (eg, rhabdoviruses) or naked (eg, picornaviruses) viruses. Viruses can also be divided according to their morphology, which can be polymorphic or structured, the latter having either icosahedral or helical symmetry.
To give an example of the taxonomic division of viruses: the 2007 pandemic H1N1 virus belongs to the species of influenza A virus, the genus of influenza virus, the family of Orthomyxoviridae with a negative-stranded ssRNA genome covered by a helical envelope. A list of common human viruses and additional viruses repeatedly referred to in this text and their classification is shown in
Table 39.1. Other characteristics of virus families, such as their genomes and surface structures, are shown in
Table 39.2.
Virus Transmission
Viruses have a single-track mind; their only goal is to replicate. The goal of their unwilling hosts is to get rid of them by mounting an immune response. Viruses have evolved a multitude of mechanisms to evade this immune response, and their hosts have adapted countermeasures accordingly, as only those that survived the onslaught of infections reached reproductive age. Highly contagious viruses can afford to not be overly concerned if their hosts, which are essential for their replication, survive, die, or mount a rapid and successful immune response because their ease of transmission ensures their continued existence. A typical example for such a virus is influenza virus, which is transmitted by aerosols before its host becomes sufficiently ill to seek solace in bed, which would limit contact with others and thus reduce the chance for the virus to spread. An example for a virus that is not overly contagious is rabies virus; it is transmitted by the bite of an infected animal, and it ensures its transmission by literally driving its host into an insane rage so that it will randomly attack and bite everyone in sight. Other viruses evolved to ensure the continued survival of their hosts, which enables their own continued replications without necessitating rapid transmission to a new individual. Such viruses are usually more complex, as much of their genome is devoted to combat immune responses.
Many viruses can only replicate in one species and therefore human infections require human-to-human contact. For viruses that are heat labile, such contact needs to be close, while more stable viruses can remain on surfaces or in water until the opportunity for infection arises. Viruses that only replicate in humans can potentially be eradicated once a vaccine becomes available as exemplified by smallpox virus. Other viruses are less discriminatory, and they replicate in multiple species, not necessarily only mammals but also birds or invertebrates. For example, influenza viruses can infect aquatic birds, chicken, swine, horses, humans, and even cats. Although they are most commonly transmitted to humans from other humans, spread from infected animals can occur, such as infections of humans with pathogenic H5N1 from chickens or ducks. Some viruses, such as rabies virus, infect all warm-blooded mammals, while other viruses alternate between mosquitoes and vertebrate animals. An example for the latter is Japanese encephalitis virus, which, in addition to mosquitoes of the Culex tritaeniorhynchus species, can infect humans, birds, most domestic animals, snakes, and frogs. The host range of a specific virus affects the mode of transmission, which is further influenced by the tissue tropism of the virus and its resistance to environmental factors like temperature or water.
One of the main protections against virus invasion is healthy skin; the upper layer of the keratinized epidermis effectively prevents entry of viruses. Mucosal surfaces,
although they are commonly bathed in antiviral proteins present in saliva and tears, in acids found in the female outer genital tract or the stomach, or in destructive enzymes such as those present in the upper intestinal tract, provide a more permissive port of entry for viruses and, consequently, most viruses are transmitted through the mucosal surfaces of either the airways, the intestines, the genital tract, or the eye. Influenza viruses, parainfluenza viruses, some types of adenoviruses, and rhinoviruses spread through the airways and are transmitted by aerosolized droplets expelled by coughing or sneezing.
Viruses that spread through aerosols tend to be highly contagious, such as influenza viruses, which cause annual epidemics and occasional pandemics that within a few months can spread throughout the world as was shown in the 2009 swine flu pandemic. The new pandemic influenza virus was first identified in Mexico on March 18th, 2009; reached California by March 28th; was detected in Canada, New Zealand, the United Kingdom, Israel, and Spain by April 28th; in Germany by the 29th; in Austria, Switzerland, and the Netherlands by April 30th; in other European countries, as well as in Asia, by May 2nd; in South America by May 5th; and on June 11th was officially declared as a pandemic virus by the World Health Organization. The total death toll of this pandemic was rather modest with approximately 5700 reported deaths by August 10th, 2010, when the World Health Organization announced the official end of the pandemic. Another highly contagious virus is varicella virus, which causes chickenpox. This virus is also spread by droplets from person to person, but, unlike influenza virus, which is fairly stable and can thus infect individuals that touch an infected surface and then their nose, varicella virus is very heat labile and, as a rule, requires direct person-toperson contact.
Other viruses are transmitted by oral ingestion and are then spread by shedding into feces. These viruses are generally stable, allowing them to resist the acidic environment of the stomach or the digestive enzymes of the intestinal tract. Many of them, such as influenza viruses that predominantly infect aquatic birds through the oral route or rotaviruses that cause severe diarrheal disease in children, can also survive for a prolonged time in water. Improperly treated drinking water can spread a number of other viruses, such as enteric adenovirus, calicivirus, astrovirus, poliovirus, or hepatitis A virus.
Sexually transmitted viral infections include herpes simplex virus (HSV) type 2, HIV-1, and several types of human papilloma viruses (HPV). Interestingly, all of these viruses establish sustained infections. HSV-2, after a replicative phase, persists latently in root ganglia from where it is periodically reactivated causing local sores that shed virus. HIV-1 first causes an acute flu-like infection and then persists mainly in CD4+ T cells while constantly dodging a vigorous antiviral immune response through mutations and immune evasion strategies. Although oncogenic types of HPV such as HPV-16 or -18 are commonly eliminated after genital infection, their persistence can over time cause transformation of the infected cells due to the activity of the two viral oncoproteins E6 and E7, which disrupt key cell cycle checkpoints and then lead to cervical cancer in women or penile or anal cancer in men. Although some of the sexually transmitted viruses (eg, HSV-2) are highly contagious, others (eg, HIV-1) transmit poorly and the average rate of HIV transmission has been estimated at 0.0082 per coital act in humans without comorbidities.
6Some viruses literally need to be injected into the body to cause an infection. These viruses are either transmitted by blood sucking insects or animal bites. Three flaviviruses, Dengue virus, West Nile virus, and Japanese encephalitis virus, are spread by mosquitoes, whereas Kyasanur forest disease virus, another flavivirus, is spread by ticks. Rabies virus, another vector-borne virus, is generally transmitted by the bite of an infected animal, most often a dog. The virus replicates in the central nervous system and is then transported to peripheral organs such as the salivary glands from where it is secreted into the saliva ready to spread to its
next victim. Although the vast majority of rabies infections are caused by bites, mucosal transmission
7 and transmission by transplantation of tissues from an infected individual have been reported.
8
Virus Cell Entry and Replication
Most viruses enter cells upon binding to a receptor, some of which are broadly expressed while others are specific for a certain cell type. In some instances, viruses bind with high avidity to one receptor but are also capable of infecting cells that lack expression of the high-affinity receptor through low avidity binding to an alternative molecule. Other viruses require binding to a receptor and a coreceptor. Receptor usage determines tissue tropism of many viruses and in some cases it also influences their host range.
For example, the hemagglutinin (HA) of influenza A viruses that can spread in humans binds to sialyated glycan receptors with a terminal α2-6 linked N-acetylneuraminic acid. In contrast, α2-3 linked sugar residues are used as receptors for influenza A viruses that circulate in birds. Once the HA has bound to its receptor, it is cleaved. A trypsin-like enzyme present only in the lung cleaves HA into two subunits, which allows the virus envelope to fuse into the cell membrane. Some of the more pathogenic strains, such as the 1918 H1N1 virus or pathogenic 2006 H5N1 viruses, activate HA through a trypsin-independent mechanism.
9 These strains have a multibasic cleavage site that can be digested by furin and furin-like proteases, which are more ubiquitously present in human tissues than the trypsin-like enzymes, allowing these viruses to infect tissues other than lung.
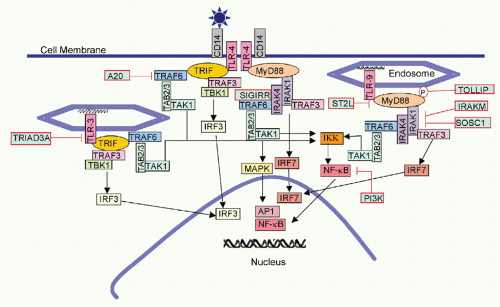
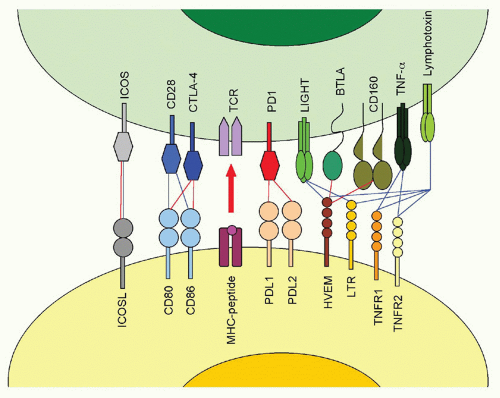
The fiber knob of adenoviruses preferentially binds the coxsackie adenovirus receptor, which is expressed on epithelial cells. In addition, an Arg-Gly-Asp (RGD) motif present within the viral penton can bind α(v)-integrins with lower avidity. The fiber of adenoviruses of subfamily B2, on the other hand, binds CD46, a ubiquitously expressed complement component that also facilitates entry of measles virus and human herpesvirus (HHV)6. The herpes virus mediator (HVEM) is a bimodal switch that can provide both immunostimulatory and immunoinhibitory signals to the immune system. Upon binding to LIGHT (lymphotoxin [LT]-like, exhibits inducible expression and competes with HSV-1 glycoprotein D [gD] for HVEM), HVEM submits stimulatory signals. Upon binding to the B- and T-lymphocyte attenuator (BTLA), it acts as an immunoinhibitor. HVEM also binds HSV-1 gD, thus facilitating entry of this virus. Binding of gD to HVEM takes place on a site that overlaps with the BTLA binding site; therefore, gD can be used to inhibit an immunoinhibitory pathway. HSV-1 may have evolved to block such a pathway, as activation of NF-κB promotes viral replication and assists in transcription of some of the early viral genes.
10The envelope protein of HIV-1 binds cluster of differentiation (CD)4 expressed mainly on T-helper cells. Upon binding, the protein undergoes structural changes that allow for its binding to a coreceptor, which for transmitting virions is CCR5, but following mutations, viruses circulating in an organism can also use CXCR4.
11 A mutant allele of CCR5 termed CCR5d32, which results in lack of CCR5 expression on the cell surface and which is found in 10% of Caucasians of European descent, provides resistance to infections with HIV-1.
12 Other viral receptors include the nicotinic acetylcholine receptor for rabies virus, heparan sulfate for dengue virus, adeno-associated viruses, and some of the herpes viruses, CD155 for poliovirus, CD81 for hepatitis C virus, CD21 for Epstein-Barr virus (EBV), C-type lectins, such as dendritic cell (DC)-specific intercellular adhesion molecule-3-grabbing nonintegrin (DC-SIGN) and liver/lymph node-specific intercellular adhesion molecule-3-grabbing integrin for Ebola virus, integrin-β3 for Hantan virus or intercellular adhesion molecule (ICAM)-1 for rhinoviruses. Examples for viral receptors including their physiologic functions are listed in
Table 39.3.
Upon binding to a receptor, viruses not only need to gain access into the cell, but most of them then have to traverse to the nucleus to initiate their replication. Viruses enter the cell either through endocytosis
31 or fusion.
32 Clathrin-mediated endocytosis is used by enveloped as well as nonenveloped viruses including adenoviruses, influenza viruses, poxviruses, or rabies virus. In cadherin-mediated endocytosis, the virus-receptor complexes cluster into a cadherin-coated pit on the cell membrane that becomes invaginated, eventually closes, and detaches from the cell membrane. The clathrin-coated vesicles then deliver their cargo to early endosomes from where it travels to late endosomes. Other viruses such as coxsackie B virus, respiratory syncytial virus (RSV), and others enter cells by caveolar endocytosis. Caveolae are invaginations in the plasma membrane that are rich in cholesterol, glycosphingolipids, and claveolin, which are used for uptake of macromolecules into endosomes. In addition, caveolar endocytosis allows for transcytosis of molecules from the basal to the apical side of a cell or vice versa. Human enterovirus has been described to enter cells through a lipid raft dependent pathway, rotavirus infects through a cholesterol- and dynamin-dependent but clathrin-and caveolae-independent pathway, while other viruses enter cells by micropinocytosis or phagocytosis.
Enveloped viruses such as paramyxoviruses, some herpes viruses, or HIV-1 invade cells by direct fusion of the virus envelope with the cell membrane. Fusion is promoted by hydrophobic sequences within a viral surface protein and causes release of the viral genome into the cytoplasm.
Viruses that enter cells through endocytosis end up in endosomes. Mechanisms of escape from endosomes differ for enveloped and nonenveloped viruses. The decrease in pH between early and late endosomes favors conformational changes of viral surface proteins by exposing their hydrophobic residues, which allow for fusion of the viral envelope with the endosomal membrane. This in turn permits escape of the viral core or the genome into the cytoplasm. Nonenveloped viruses disrupt the endosomal membrane either by a pathway called carpet mechanism or by forming pores. In carpet-like disruption of endosomal membranes, viral peptides act like a detergent and thus interrupt the hydrophobic interactions between membrane lipids allowing for the development of micelles and for transient formation
of holes within the membrane.
33 Other viruses carry proteins, which form amphipathic α-helices that assemble into a pore within the lipid membrane of the endosome where the hydrophobic parts interact with the lipid bilayer while the hydrophilic parts of the coils form the inner wall of the pore.
34 Picornaviruses, parvoviruses, and reoviruses utilize this strategy.
Once within the cell, viruses may be broken down by autophagy,
35 a catabolic process involving the degradation of a cell’s own components to convert unneeded pieces into nutrients. During this process, so-called autophagosomes form from membrane structures containing autophagiarelated gene products (Atg), such as the ubiquitin-like Atg8, the Atg4 protease, and the Atg12-Atg5-Atg16 complex. The outer membrane of the autophagosome fuses with a lysosome to allow for degradation of its contents. Formation of autophagosomes is initiated by PI3K and Beclin-1. Most viruses block this pathway by inhibiting PI3K activation, but rhinoviruses and poliovirus sponsor formation of early autophagosomes but block their fusion with lysosomes and then use the structures to egress the cells.
36Once a virus has reached the cytoplasm, it must deliver its genome to the nucleus. Many viruses such as herpesviruses and adenoviruses use microtubules to reach nuclear pores. Very small genomes can diffuse passively though pores into the nucleus, while larger genomes or particles require an energy-dependent process. Some viruses use viral proteins to facilitate nuclear entry. For example, cytomegalovirus (CMV) encodes two proteins, pUL69 and pUL84, that facilitate the transport of its genome to the nucleus. pUL69 binds to UAP56, which facilitates nuclear export of unspliced RNA; pUL84 binds to importin-alpha proteins,
37 which can dock to nuclear pores and then be transported through it.
Most viruses initiate their replication in the nucleus as they depend on nuclear enzymes for transcription. Poxviruses and some of the RNA viruses are independent of such enzymes and can replicate their genome in the cytoplasm. HIV-1 replicates in the nucleus after it reverse transcribes its RNA in the cytoplasm.
Replication of different types of virus can be exemplified using the following viruses: adenovirus, a dsDNA virus; adeno-associated virus, an ssDNA virus; reovirus, a dsRNA virus; poliovirus, a positive-sense ssRNA viruses; influenza A virus, a negative-sense ssRNA virus; HIV-1, a positive-sense ssRNA-RT virus; and hepatitis B virus, a dsDNA-RT virus.
Adenovirus transcription is typical for that of some of the larger DNA viruses as it proceeds in stages. Initially, the immediate early gene is transcribed. The resulting gene products alter the host cell to provide a more favorable environment for viral replication and initiate transcription of early viral genes, which have regulatory functions and serve to modify host cell functions or subvert immune responses. Thereafter, the viral genome replicates concomitantly with transcription of the late viral genes that encode
structural proteins. Specifically, the replication cycle of adenovirus starts with expression of E1A, which encodes two polypeptides that bind to cellular proteins, including cellular transcription factors, which in turn changes the cell’s gene expression profile and allows for transcription of the other early viral genes E1B, E2, E3, and E4. E1A promotes apoptosis, while E1B proteins are antiapoptotic. E1B polypeptides turn off host cell protein synthesis and help to stabilize, transport, and selectively translate viral RNA. E2 encodes DNA-binding proteins and a polymerase. E3 gene products are nonessential for virus replication but serve to evade immune responses. E4 encodes seven polypeptides, which collaborate with E1 gene products in promoting viral transcription and modulating host cell functions. E4 gene products are also essential for nuclear export of viral RNA. Adenoviruses also encode one or two virus-associated (VA)-RNA species, which form short hairpin loop structure of approximately 200 bases, are transcribed by polymerase III and stimulate translation of viral genes. VA-RNA can be processed into shorter RNAs and act as micro RNA,
38 inhibiting activation of protein kinase R, which inhibits further messenger RNA (mRNA) synthesis through phosphorylation of the translation initiation factor EIF2A. Transcription and translation of early gene products is followed by DNA replication, which is initiated by a terminal protein that is covalently bound to the 5′ ends of the long terminal repeats. Once DNA replication is initiated, the late gene products, which form the viral capsid, are produced from the L1-L4 domains. Viral assembly begins in the cytoplasm and is completed in the nucleus.
Adeno-associated viruses are ssDNA viruses that cause no known disease in humans. They are dependoviruses and require coinfection with another virus, most commonly an adenovirus, to complete their lifecycle. The approximately 4.7 bp genome is flanked by terminal repeats that contain a multipalindromic terminus that forms a loop and thereby promotes priming for DNA replication. The genome contains only two genes; one, the rep gene, encodes four regulatory proteins needed for DNA replication and conversion of the dsDNA intermediate into the final ssDNA. The viral capsid is composed of three virus proteins derived from the cap gene by transcript splicing. Initiation of transcription of the rep gene requires proteins from a helper virus such as gene products from E1, E2, E4, as well as VA-RNA from adenovirus. Final assembly of the adeno-associated virus takes place in the nucleolus.
Reovirus infections are asymptomatic in humans but cause disease in newborn mice. This virus, which contains 10 to 12 segments of dsRNA, replicates in the cytoplasm of infected cells without completely uncoating. RNA is transcribed from the negative strand of the genomic RNA and leaves the capsid to be translated. Secondary transcription occurs later followed by assembly within the cytoplasm.
The viral genome of positive-sense ssRNA viruses, such as poliovirus, a picornavirus, can directly serve as mRNA. Poliovirus RNA lacks the methylated cap structure that is typical for mammalian mRNA but rather has an internal ribosomal entry site. To avoid competition with translation of mammalian mRNA, poliovirus interferes with recognition of the host’s methylated cap, thus inhibiting host cell protein synthesis.
39 The poliovirus RNA is translated into a single polypeptide that is cleaved into a replicase, proteases, and structural proteins. The polymerase transcribes the positive-stranded RNA into a minus-sense RNA to serve as template for new positive-stranded RNA. The latter can either be translated, serve as template for minus stranded RNA, or be packaged into new virions. Replication as well as assembly occurs in the cytoplasm.
Influenza viruses are segmented negative-sense ssRNA viruses, which replicate in the nucleus. The RNA-dependent RNA polymerase transcribes positive-stranded RNA segments that are either transported to the cytoplasm for translation or remain in the nucleus to serve as templates for negative-stranded RNA synthesis. Newly produced internal proteins are transported into the nucleus where they, together with RNA segments, form new virus particles. The two viral surface proteins, the HA and the neuraminidase are secreted through the Golgi apparatus to the cell surface where they are then picked up by the envelope once the virus leaves the cell.
HIV-1 is initially reversed transcribed in the cytoplasm by the viral reverse transcriptase into an RNA/negativestranded DNA hybrid. This process is error prone and contributes to the high mutation rate of HIV-1. The RNA is degraded, and a positive-stranded DNA is synthesized allowing for the formation of a dsDNA, which, together with some enzymes, enters the nucleus; there the viral genomes integrates with the help of the viral integrase and serves as a template for synthesis of viral transcripts. Two newly produced viral proteins, Tat and Rev, are essential for efficient protein production: Tat by enhancing transcription and Rev by supporting export of unspliced mRNA from the nucleus, which allows for production of the structural proteins Gag and Env. The full-length viral RNA binds initially to Gag and is then packaged into new virus particles. Env is transported to the cell surface after it is cleaved into two subunits and, with the help of cellular chaperone proteins, folded into a trimer. Assembly of mature virions takes place at the plasma membrane.
Hepatitis B virus (HBV) carries a circular partially dsDNA genome that encodes four structural and two nonstructural proteins through overlapping open reading frames (ORFs). Within the nucleus of an infected cell, the genome is converted into a full dsDNA, which serves as a template for the viral transcripts. The largest mRNA, which is longer than the viral genome, is called the RNA pregenome and is packaged into core particles within the cytoplasm. Within these particles, the pregenomic RNA is reverse transcribed into viral DNA genomes. Upon synthesis, the viral surface protein is transported to the cell membrane and complete assembly of the virion takes place during budding of the virus.
Once replication is completed and full virions have been assembled, viruses need to leave the cells. This again can occur through several pathways. Some viruses, such as HIV-1 or influenza virus, assemble their newly synthesized viral surface proteins on the cell surface and then bud through this part of the cell membrane, picking up not only their own surface proteins but also membrane proteins belonging to
the host cell. Budding eventually destroys the cell membrane and leads to cell death. Other viruses, especially those that are nonenveloped, instruct the infected cell to undergo apoptosis and virus released from dying cells is encapsidated into apoptotic bodies, which are taken up by neighboring cells, thus facilitating infection of new cells. Some viruses are released by exocytosis, a process that resembles reversed pinocytosis in which virus particles are encapsidated into small vesicles that enter the secretory pathways. This form of exit does not kill the cells and is used by so-called nonlytic viruses.