SURFACE STRUCTURE OF GRAM-POSITIVE AND GRAM-NEGATIVE BACTERIA
Differences among bacteria contribute to their specific adaptation to either a particular host species or to microenvironments within their host. In general, we currently have only a limited understanding of the limitations in nutrients and the specificity of enzymes and bacterial adhesins, or host receptors, that account for these highly specific microbial tropisms. The diversity of bacterial structures encountered by the host offers a major challenge to immune detection of potential pathogens. There are no structural features that reliably differentiate pathogens from nonpathogens. Moreover, many important extracellular pathogens exist mainly as commensals, only causing damage when the balance between host and microbe is perturbed (opportunistic pathogens). Thus, their diversity requires that initial recognition through innate immunity be focused on the more conserved structural features or “molecular patterns” of bacteria. These molecular patterns are, in general, structures such as peptidoglycan, lipoteichoic acid (LTA), and lipopolysaccharide (LPS) that are also essential for bacterial viability and are, thus, unlikely to be modified so as to evade innate immunity. A further consideration is that these structural differences between types of bacteria may dictate differences in host responses and contribute to distinct patterns of disease. That said, different types of bacteria may also give rise to remarkably similar host responses. The syndrome of sepsis, for instance, looks very similar regardless whether it is caused by a gram-positive or gram-negative organism, although there may be little overlap in the specific bacterial mediators involved.
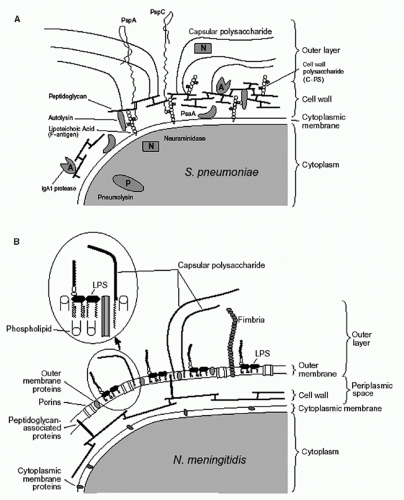
Most extracellular, as well as intracellular, pathogenic bacteria can be divided into two major groups (gram-negative and gram-positive) based on their response to staining with Gram stain. To illustrate the surface of the bacteria in the two groups, the surface structures of
Streptococcus pneumoniae (Panel A) and
Neisseria meningitidis (Panel B) are shown in
Figure 41.1. Three layers are commonly recognized: cytoplasmic membrane, cell wall, and outer layer. Although these layers are described in detail in the following, it is important to note that these definitions are operational and that, in reality, the layers are not entirely distinct. For instance, molecules anchored in the cytoplasmic membrane or cell wall may extend into or through other layers. It is also important to note that the capsule, O antigens, and cell wall are not contiguous shields; rather they are permeable enough to allow through secreted products and nutrients as well as some immunologic factors (eg, antibodies and complement).
All bacteria have a cytoplasmic membrane, a non-sterol-containing phospholipid bilayer. This membrane is an osmotic barrier and also forms a barrier for most molecules. The cytoplasmic membrane has various proteins, many of which function in transport. Some of these proteins, referred to as lipoproteins (eg, pneumococcal surface adhesion A, which is a manganese permease in
S. pneumoniae), are noncovalently anchored to the membrane through
lipid modifications and are especially common among some bacteria (eg,
Borrelia burgdorferi and
Mycobacterium tuberculosis). Proteins not exposed on the surface generally display a greater degree of structural and functional conservation compared with those exposed to the selective pressure of host immunity.
A cell wall is found in all of the pathogenic bacteria of both groups, with the exception of mollicutes (which include the genus
Mycoplasma). The cell wall surrounds the cytoplasmic membrane and is made of peptidoglycan, which is a polymer of alternating sugars N-acetylglucosamine and N-acetylmuramic acid, the latter being connected to a stem peptide. The stem peptides include four to five D- and L-amino acids that are extensively cross-linked by bridges that provide rigidity to the cell wall and protect it from environmental extremes (especially differences in osmolarity). These cell wall peptides include atypical amino acids such as diamino pimelic acids, which are the anchoring site for Braun lipoprotein of gram- negative bacteria and are found in most gram-negative bacteria but in few gram-positive bacterial species. Peptidoglycan polymerization is carried out by enzymes, many of which are the target of β-lactam antibiotics and are referred to as penicillinbinding proteins. Compared with gram-negative bacteria, gram-positive organisms may have different stem peptides and cross-linking as well as a thicker (20 to 30 nm compared with 2 to 4 nm) cell wall layer that can retain the Gram stain better. The thick cell wall of the gram-positive bacteria may be responsible for their greater resistance to complementmediated lysis. Other features of the cell wall such as the O-acetylation of N-acetylmuramic acid or the deacetylation of N-acetylglucosamine
5,6 found in some species mediate resistance to lysozyme, an enzyme that lyses bacteria by cleaving the peptidoglycan backbone.
6In addition to peptidoglycan, many gram-positive bacteria have polysaccharide (PS) associated with their cell walls, with this cell wall PS often extending into the outer layer. The structure of the cell wall PS of gram-positive bacteria varies between species but is relatively invariant within a species. Differences in the antigenicity of the cell wall PS have been used to distinguish species (eg, separate streptococci into groups A, B, C, etc.).
7,8 Cell wall PS often has phosphate group(s) in repeating units of glycerol or ribitol in a structure known as teichoic acid. Teichoic acid may also be linked to lipid molecules and is then called LTA, which is anchored to the cytoplasmic membrane and extends out through the cell wall.
9 In pneumococci, the overall PS structures of LTA and cell wall teichoic acid (also referred to as C-polysaccharide) are very similar, with the difference being their mode of attachment to the bacterial surface.
10,11Another major difference between the surface structures of gram-negative and gram-positive bacteria is the presence of an outer membrane on gram-negative bacteria. The outer membrane contains many proteins, including channel-forming porins. The outer membrane is an asymmetrical lipid bilayer. The inner leaflet is comprised primarily of phospholipids while the outer leaflet contains lipid A, the hydrophobic component of LPS. LPS, also called endotoxin, is an amphipathic glycolipid with four distinct regions: lipid A, the inner core, the outer core, and, in some species, the O antigen. Lipid A is composed of a dihexosamine backbone to which between five and seven saturated (12- to 16-carbon) fatty acids are attached through amide and ester linkages. Lipid A is the principal “toxin” associated with most gramnegative bacteria, although it is now clear that lipid A is not a true toxin. Rather, its ability to induce cytokines accounts for its potentially detrimental effects. The carbohydrate portion of the LPS, which makes a minimal contribution to its endotoxin activity, is attached to lipid A through a molecule unique to gram-negative bacteria called ketodeoxyoctanoate. Together with heptose moieties, this molecule forms the inner core of the LPS.
The outer core is composed of 7 to 10 monosaccharide units whose arrangement is relatively conserved among gram-negative species.
12 In many gram-negative bacteria, the outer core of LPS is connected to a repeating series of carbohydrates called the O antigen. The O antigen forms a hydrophilic shield around the bacterium that provides a barrier to complement deposition on the bacterial cell surface. The O antigen is variable in length, is antigenically diverse, and confers serotypic specificity. The O antigens of
Escherichia coli,
Klebsiella, and
Salmonella have as many as 30 repeating units composed of four to six sugars each.
12 Members of the genera
Neisseria and
Haemophilus, on the other hand, lack LPS with O antigens but instead have lipooligosaccharides, which have short oligosaccharides that do not exceed 7000 daltons.
For many pathogenic extracellular bacteria, PS components dominate the outer layer. In addition to the PS on LPS (gram-negative) and teichoic acid (gram-positive), there is often another thick layer of carbohydrate referred to as “capsule” that may account for more than half of the bacterial mass. An exception to this general rule is
Bacillus anthracis, whose capsule is made of poly-D-glutamic acid rather than a polysaccharide.
13 S. pneumoniae has capsular PS that is covalently attached to the cell wall in most (but not all) serotypes.
14 In contrast, the capsule PS is anchored to the outer membrane by acyl chains in
N. meningitidis15 and
Haemophilus influenzae type b.
16 Capsular PSs may be highly diverse both within and between species. In the case of
S. pneumoniae, each strain expresses a single type of capsular PS, with members of this species being capable of synthesizing more than 90 structurally distinct types.
17,18 This diversity limits immune recognition until antibody is generated to the capsular PS of the infecting strain (antigenic variation).
The outer layer is well developed in bacteria that cause extracellular infections and has many features that help the bacteria circumvent the host immune system. First, the outer layer has properties that reduce the attachment of extracellular bacteria to eukaryotic surfaces, including those of phagocytes. Generally, the PS capsules render the bacteria hydrophilic and negatively charged like eukaryotic cell surfaces, which are rich in sialic acid. By enhancing the degradation of C3b, the negatively charged surface makes the bacteria partly resistant to the deposition of complement by the alternative pathway.
19 Second, in some cases, elicitation of antibody is minimized because the capsular PS or
LPS mimics host antigens, as is more fully described in the section on Antigen-Specific Host Defense Response of this chapter. Third, by physically masking most of the other bacterial surface components, the outer layer minimizes the number of exposed epitopes that can be recognized by the antibody and complement. Although the capsule is porous to antibodies and complement, the binding of antibodies and fixing of complement beneath the capsule surface are relatively ineffective in promoting opsonophagocytosis and clearance.
20
BACTERIAL VIRULENCE FACTORS
Extracellular bacteria often elaborate molecules called “virulence factors” that are useful to their survival and proliferation in the host. For example, the shielding function of the outer layer is further augmented by the presence of surface proteins that can interfere with host clearance mechanisms. Proteins inhibiting effective complement deposition include M-protein in
Streptococcus pyogenes,
21 pneumococcal surface protein A (PspA), and pneumococcal surface protein C, which is alternatively called choline binding protein A or the C3-binding protein in
S. pneumonia.22,23 An example of a protein that interferes with antibody is protein A, which is expressed on the surface of
Staphylococcus aureus and binds immunoglobulin (Ig) in a manner that precludes recognition of its target antigen.
24 In addition, many successful mucosal pathogens, including members of the
Neisseria,
Haemophilus, and
Streptococcus genera, express proteases with specificity for the hinge region of human IgA1.
25 These IgA1 proteases remove the Fc
α component required to promote the inflammatory process, leaving the organisms’ antigens obscured by the binding of inert Fab
α fragments. By inhibiting the deposition of complement or antibody, many of these proteins act to diminish phagocytosis.
The best-known virulence factors are toxins, which interrupt specific host functions. These proteinaceous molecules (also referred to as exotoxins to differentiate them from endotoxin) can be grouped on the basis of their molecular structure and their mechanism of action.
26 The largest group are called A-B toxins, which are comprised of two subunits, each with a different function. The A subunit has enzymatic activity, and the B subunit targets the A subunit to the host cells. This group includes diphtheria toxin, cholera toxin, pertussis toxin, and two anthrax toxins (lethal factor and edema factor). For instance, the lethal factor of
B. anthracis behaves as the A subunit and requires a B subunit protein named “protective antigen” to enter into target cells. In some cases, the toxin alone is sufficient to account for the detrimental symptoms of its respective infection. Cholera toxin causes ADP ribosylation of G proteins, which stimulates adenylate cyclase and increases cyclic adenosine monophosphate in cells lining the gut. This results in the secretion of electrolytes and is responsible for a severe diarrhea, which promotes transmission of
Vibrio cholerae but often also causes dehydration that, if not treated, may be fatal. Uptake of botulism toxin by nerve endings leads to retrograde transport that interrupts synaptic transmission and causes a flaccid paralysis.
27 Staphylococcal enterotoxin A (a toxin that acts from the gut lumen), which is one of five membrane-damaging toxins produced by staphylococci, is the primary cause of staphylococcal food poisoning and plays a major role in invasive infections.
28 Some strains of
E. coli produce a protein synthesis-inhibiting verotoxin, which may damage the microvasculature of the kidney and cause hemolytic uremic syndrome.
29 Another group of proteins secreted by
S. aureus and
S. pyogenes have toxin-like effects but lack any enzymatic activity. These “superantigens” cause a nonclonal stimulation of T cells by bridging major histocompatibility complex (MHC) class II molecules (outside the antigen-binding site) on antigen-presenting cells with the Vβ region of the T-cell receptor on T cells. The ensuing massive release of cytokines by localized release of a superantigen, such as the toxic shock syndrome toxin 1 expressed by some strains of
S. aureus, results in systemic symptoms that are collectively known as “toxic shock syndrome.”
30Another class of virulence factors neutralizes host defenses. As stated previously, many virulence factors interfere with complement deposition on bacteria.
S. pyogenes and group B streptococci produce a C5a peptidase that inhibits the chemotaxic effects (recruitment of host phagocytes to the sites of infections) of C5a, a product of complement activation.
31 Pneumolysin produced by
S. pneumoniae is a member of a large class of cholesterol-dependent cytotoxins that oligomerize to form large pores, which interfere with a number of host cell functions or induce cell death when present in higher concentrations.
32 Pneumolysin also depletes complement at a distance from the pneumococci and interferes with both the function of phagocytes and the development of protective immunity.
33 Helicobacter pylori produces urease, which can generate ammonia that can neutralize acid in the stomach and thereby promotes the survival or the organism.
S. aureus produces a pigment that makes the bacterium more resistant to oxidative stress and killing by neutrophils.
34While evasion of professional phagocytes is critical for extracellular pathogens, the ability to attach to other cell types, including both mucosal and nonmucosal surfaces, is important for their persistence. Many bacterial surface proteins have an adhesive function that confers a high affinity for binding to specific host cell receptors. Nasopharyngeal carriage of pneumococci is mediated largely by adherence to the host molecules N-acetylglucosamine β1→3 galactose or N-acetylglucosamine β1→4 galactose.
35 Bacteria often mimic host ligands in order to coopt host receptors for their own purposes. Many pathogens of the airway express phosphorylcholine (phosphocholine [PC]) on their surfaces.
36 This molecule, which is otherwise unusual in bacteria, is found on platelet-activating factor (PAF) and allows bacterial binding to its receptor (rPAF).
37 To facilitate their attachment to host cells, many bacteria use pili (fimbriae), long filamentous structures extending from the organism. The Pap pilus of
E. coli binds the galactose α1→4 galactose unit of cell surface globoside in urethral epithelial cells.
38 The
V. cholerae pilus allows that bacterium to attach to the enterocyte for more efficient toxin delivery.
39,40 Bordetella pertussis has three adherence factors—a filamentous hemagglutinin,
pertactin, and a pilus—that allow it to attach to ciliated respiratory epithelial cells in the trachea and bronchi and thus resist the cleansing action of mucus flow.
41,42Another group of virulence factors helps bacteria acquire essential nutrients. Motile bacteria (eg,
Pseudomonas aeruginosa) express flagellin, a complex motor apparatus that allows the bacterial cell to transit along a concentration gradient of nutrients.
43 While in some cases the host and microbe provide nutrients for one another, for other nutrients there is fierce competition. Mucosal fluid and blood are low in free ferric iron due to the presence of iron-binding proteins such as lactoferrin and transferrin. To successfully compete with the host for this vital metabolite,
N. meningitidis,
Neisseria gonorrhoeae, and
H. influenzae have complex surface transport systems that can obtain iron from human transferrin, lactoferrin, and hemoglobin.
44 Other bacteria such as
E. coli and salmonella use a different mechanism to acquire iron; they secrete small, high-affinity iron chelators, called siderophores, that remove iron from human proteins in the environment surrounding the bacteria. The iron-siderophore complex is then taken up by the bacterium, which then degrades the siderophore so that the iron can be freed for its use.
45Production of virulence factors is often highly regulated by bacteria in order to adapt to different environments, such as the natural environment outside of a host, the mucosa of a host, or more invasive sites within a host. For staphylococci, it has been shown that the amount of capsule is regulated in response to environmental stimuli.
46 One of the best studied of such regulatory systems is the BvgAS, a two-component regulatory system in
B. pertussis.47 This system, which regulates the expression of adhesins, toxins, and other virulence factors, is controlled by external signals including Mg
2+, temperature, and nicotinic acid. Two proteins are involved in this regulatory system: BvgS and BvgA. BvgS, the sensor, is a kinase and is able to autophosphorylate itself in response to the environmental signal. BvgA, the response regulator, is in turn phosphorylated by BvgS. Phosphorylated BvgA is able to activate transcription of virulence genes through a change in its interaction with a 70-bp consensus sequence repeated in bvg-regulated promoters.
47 Analogous two-component regulatory systems in other pathogens are frequently used to regulate the expression of genes associated with virulence.
48Another strategy used by extracellular pathogens depends on selection among a heterogeneous population for those members with permissive characteristics. This heterogeneity in a population is commonly generated through genomic rearrangements, such as recombinational events or slipstranded mispairing in highly repetitive deoxyribonucleic acid (DNA) sequences.
49 This latter mechanism allows for reversible on-off switching (phase variation) and is especially prevalent in genes encoding cell surface structures subject to immune pressure. For instance, the capsular PS on
N. meningitidis is needed to protect the organism during invasive infection but inhibits adherence on the mucosal surface where complement is less abundant. Phase variation of a gene required for capsule synthesis allows for selection of organisms without capsule (phase-off).
50 This change facilitates the bacterial adhesion to the epithelial cells, perhaps by exposing the bacterial adhesins. Alternatively, by decreasing capsule production, the bacteria become less hydrophilic and less negatively charged. This change facilitates their entry into the epithelial cells and their subsequent invasion into deeper tissues. Upon the emergence of the bacteria from the epithelial cells into the submucosa, capsule synthesis is restored (phase-on) because of the selective pressure of complementmediated clearance and the requirement for the capsule to survive where the concentration of complement is higher. The flexibility to express different surface properties helps bacteria successfully evade the host immune system and survive in many niches within the host.
Bacteria-to-bacteria signaling is another important mechanism for the control of virulence factors. This phenomenon, called “quorum sensing,”
51 has been shown to be operative in a large number of gram-negative and grampositive species. The signal transmitted between the bacteria can be an acylated lipid (eg, homoserine lactone) in gramnegative bacteria (eg, vibrios) or a peptide in gram-positive bacteria (eg,
S. aureus). Quorum sensing has been shown to be important in biofilm formation in a number of bacterial species, in the expression of “competence” for the uptake and incorporation of exogenous DNA (transformation), and in the regulation of a number of virulence factors.
52 Biofilms are communities of one or multiple bacterial species that adhere both to each other and to a target surface. Bacteria in biofims are particularly resistant to many host clearance mechanisms and to antibiotics that are effective against free-living (planktonic) bacteria. Biofilms, therefore, are often a contributing factor in more chronic bacterial infections such as those involving foreign bodies or chronic otitis media.
53An important characteristic of virulence factors is their structural polymorphism. For instance, there are at least 100 different serologic types of M proteins of
S. pyogenes.
54 Similarly, pneumococci have more than 90 serologically distinct capsular PSs.
17,18 The polymorphism in the structure of many virulence factors allows the bacteria making them to avoid antigen-specific host immunity. For example, antibodies to the immunodominant region on one serotype of M protein do not cross-react with M proteins of other serotypes and thus do not provide protection against strains expressing other serotypes. Similarly, newly acquired pneumococci can escape recognition by anticapsular antibodies produced in response to previous pneumococcal infections with other serotypes.
The polymorphism in virulence factors is achieved by various genetic mechanisms. Variation in M proteins is the result of sequence differences in the N-terminal (but not C-terminal) half of M proteins.
55 S. pneumoniae has the genes for synthesizing capsular PS as a “genetic cassette” that can be exchanged among different strains
56 and may result in the shift in the serotype distribution following the use of vaccines designed to elicit serotype-specific protection.
57,58 Neisseria has genetic machinery for rapid gene rearrangement
59 through gene conversion using the multiple “silent” pili genes with different sequences. This process, similar to gene rearrangements that generate specific Ig, permits an individual bacterium to quickly produce progeny expressing pili with different characteristics. The number of potential
pilus-antigen variants within the progeny of a single organism is estimated to be greater than 100,000.
60 In addition,
Neisseria contain large numbers of genes with tandem repeats that undergo phase variation through slip-stranded mispairing of these sequences. Based on predictions from whole genome sequencing, through this mechanism alone the organism may be able to generate more than 2
65 different phenotypic variants.
61Sequencing of the entire genomes of bacteria has shown that the genes for virulence factors have generally originated from other organisms and exist as a part of large blocks of DNA containing multiple genes. These DNA blocks are called “pathogenicity islands” (PAIs). For instance, strains of “enteropathogenic”
E. coli contain a PAI encompassing about 41 genes encoding a surface ligand required for intimate association of the bacterial and host cells and for a bacterial secretion apparatus.
62 This system (type IV secretion system) allows for delivery of the receptor for its own adhesin, encoded on the same PAI, into host cells. Elaborate secretion mechanisms (types III and IV) and pore-forming toxins are now known to be mechanisms whereby extracellular organisms gain access to the host cell cytoplasm to modify its activity to suit their needs. Another example is
H. pylori, which injects cytotoxin-associated gene A (CagA) molecules directly into host cells using a syringe-like type IV secretory apparatus. CagA is then phosphorylated by the host cells, and the phosphorylated CagA alters host cell function, with the
H. pylori strains producing the CagA molecules being more likely to cause ulcers.
63 In the case of
S. pyogenes, its pore-forming toxin, streptolysin O, allows for translocation of an enzyme (NAD-glycohydrolase) that is capable of producing the potent cytoplasmic second messenger, cyclic ADP-ribose.
64
BACTERIAL INVASION OF THE HOST
Both keratinized skin and mucosal surfaces have inherent nonimmune defense mechanisms that modulate bacterial growth and minimize the risk of invasion. Healthy human skin is an effective physical barrier to infection by most human extracellular and intracellular pathogens. The keratinization of fully differentiated skin epithelium results in a relatively impermeable surface. In addition, lysozymes, toxic lipids, and hydrogen ions secreted by cutaneous glands offer bacteriostatic protection for cutaneous pores and hair follicles. Occasionally, this defense can be breached by extracellular bacteria such as
S. pyogenes or
S. aureus, causing cellulitis and abscess. More commonly, bacterial invasion through intact skin requires physical damage, such as abrasions, burns, or other trauma. For instance, cutaneous anthrax develops when
B. anthracis enters the body through a break in the skin.
Staphylococcus epidermidis, a member of the commensal skin flora, can infect indwelling catheters by invading through the puncture site in the skin and may lead to bacteremia or colonization of prosthetic devices, including artificial heart valves and shunts. A major factor allowing these bacteria to cause disease is their ability to form a biofilm, which facilitates their adhesion, is antiphagocytic, and acts as a barrier to antibiotic penetration.
65Unlike the skin, the mucosal epithelium is not keratinized. Instead, mucosal areas, such as the gastrointestinal tract, nasopharynx, upper airway, and vagina, are moist and nutritionally rich. Thus, it is not surprising that mucosal areas contain a large number of bacteria. In oral secretions and gastrointestinal products, 10
8 and 10
11 bacteria/mL may be found, respectively. To ensure their survival in the mucosal environment, extracellular bacteria elaborate many virulence factors required for the acquisition of essential nutrients or for their adherence to the host cells. In some cases, bacteria may subvert the host inflammatory response. Salmonella species can block the activation of NF-κB and the subsequent activation of the inflammatory response. They achieve this by preventing the degradation of IκB, which is essential for the translocation of NF-κB from the cytoplasm to the nucleus.
66 In some cases, pathogens locate in a less well-protected microenvironment within the mucosal areas.
H. pylori survives in the very acidic stomach by burying itself in the mucus, which protects it from direct exposure to the acid and from phagocytes. There are also few other species for
H. pylori to compete with in this more hostile environment.
Mucosal sites play host to an especially diverse array of bacterial species. Most of the bacteria species found at mucosal sites are harmless. In addition, polymerase chain reaction analysis of 16S ribosomal ribonucleic acid sequences suggests the presence of many additional unidentified (and so far unculturable) bacterial species on the mucosal surface.
67 Many potentially pathogenic bacterial strains are also often found in the mucosal areas of healthy individuals without causing symptoms.
S. pneumoniae,
N. meningitidis,
H. influenzae, and
S. aureus are examples of pathogenic extracellular bacteria that are frequently carried in the nasopharynx of healthy individuals. The carriage rate of pathogenic bacteria can be relatively high; for example, 50% to 60% of healthy young children may carry
S. pneumoniae in their throats.
68Maintenance of species diversity in mucosa is dynamic. In some situations, collaboration among bacteria is essential for their successful colonization, as seen among the complex hierarchical communities adhering to tooth surfaces. In other situations, bacterial species compete and regulate diversity among themselves.
36 Many bacterial species produce molecules that suppress the growth of other bacterial species. These molecules may include bacteriocins, small molecules that target members of the same or different species that do not express the same bacteriocins.
69 Some species can take advantage of host responses to which they are resistant to outcompete another member of the same niche that is less resistant.
36 The host may also control the diversity of colonizing bacteria by modifying the pH or other environmental conditions in the mucosal area. Interference with these homeostatic mechanisms, as occurs with antibiotic therapy, may alter the flora and predispose the host to disease. As noted previously, stomach acid is an effective barrier to reaching the nutrient-rich environment of the gut. When stomach acid is pharmacologically reduced, the inoculum of organisms like
V. cholerae required to infect the intestines is greatly diminished.
Several explanations have been advanced to explain why many pathogens that colonize mucosal sites do not cause disease. One explanation is that the maintenance of diverse bacterial population is responsible for the prevention of the disease. For instance, the destruction of the normal gastrointestinal bacterial flora with some antibiotics can be associated with the selective expansion of
Clostridium difficile and the development of pseudomembranous colitis.
70 Another explanation may be that the pathogenic bacteria carried in healthy persons are different from those isolated in disease settings. For instance, during nonepidemic periods, approximately 5% to 10% of the population carries
N. meningitidis, which are mostly nonencapsulated.
71 During epidemics, 30% to 60% of the population may carry meningococci, which are mostly encapsulated and the majority of which are of the same capsular type as the case strain causing the epidemic.
72 A third explanation is that the pathogenic bacteria are effectively confined to the mucosal surface where they do not cause damage or induce inflammation. Group B streptococci are carried asymptomatically in the lower intestine and the female genital tract. In the same host, in the setting of parturition, group B streptococci may access the bloodstream and cause septic infection. A fourth explanation is centered on the differences among hosts. Group B streptococci that colonize the mother may cause life-threatening infection when the same strain is passed to the neonate at or before birth.
73Although pathogenic extracellular bacteria can exist asymptomatically in the mucosa, they can passively enter into less-well-defended sites and cause focal infections. For instance,
E. coli, normally present in the gut, may enter the normally sterile urogenital tract and cause urinary tract infections.
S. pneumoniae and
H. influenzae are often carried in the nasopharyngeal space, but they can invade nearby normally sterile cavities (eg, lungs, sinuses, and the middle ear) and cause focal infections. Aspiration of bacteria from the nasopharynx into the lungs most likely occurs frequently with no ill effects; however, aspiration may lead to an infection when there is damage to the epithelial surface, particularly when the protective effects of mucociliary activity are lost, as often occurs in a smoker or during recent viral infection (respiratory syncytial virus or influenza).
7 Some bacteria produce enzymes such as hyaluronidase,
74 which may aid in their passage through tissue barriers.
Bacteria can actively invade deeper tissues by multiple pathways. They can enter through specialized cells. Shigella, for example, can breach the gut mucosa by transcytosing through the M cells in the gut.
75 Alternatively, extracellular bacteria can breach a cellular barrier (epithelium or endothelium) by going through (transcytosis) or between (paracellular pathway) the cells.
75 Porphyromonas gingivalis, an organism associated with adult periodontitis, may breach the epithelial layer by the paracellular pathway through the production of enzymes useful in digesting the tight junction.
76 Two different mechanisms of transcytosis have been described for pneumococci. In one, pneumococci may cross the bronchial epithelial cells by binding the polymeric Ig receptor of the epithelial cells and traveling in a retrograde manner by the IgA secretory pathway.
77 In the other, pneumococci may use PC to bind to rPAF, which is abundant on activated endothelial cells, epithelial cells, or pneumocytes.
35,78 In many cases, bacterial adhesion triggers changes in the host cell function, and these changes can assist transcytosis. For instance, nontypeable
H. influenzae with LPS glycoform-containing PC can bind to rPAF on endothelial cells and initiate signaling through this receptor.
79