7.1 Introduction
Innate immunity has evolved over several billion years, and has selected recognition systems that can discriminate between molecules expressed by infectious agents and their hosts. Innate immunity rarely causes direct damage to the host. The same is not the case for adaptive immunity, which has been around for about one-eighth of the time of innate immunity and is a much less perfect system. This imperfection has arisen largely because the antigens expressed by pathogens that the adaptive immune system needs to recognize are often not inherently different from the host’s own molecules. This means that the antigen receptors of the adaptive system cannot be pre-selected to recognize only pathogen-expressed antigens. The outcome is that both the T cells and antibodies of adaptive immunity can potentially recognize host antigens. On occasion this can result in serious damage and sometimes death to the host. However, the damage is largely caused by recruiting and amplifying components and functions of innate immunity that are otherwise needed to deal with infections. In this chapter we examine the different types of immune-related diseases and other conditions that directly involve the immune system. We also introduce some types of therapy that are being used or developed to treat them, by redirecting immunity to another end and some cases by using components of immunity as therapeutic tools.
We start with a brief overview of the different areas we will discuss (Section 7.1). Then we consider the different ways in which the immune system can cause tissue damage and review what we know of the underlying causes of these harmful responses (Section 7.2). We focus on how lymphocytes are normally regulated to discriminate between infectious agents and host molecules (leading to active responses in the first case, tolerance in the second), under what circumstances they can be triggered to make abnormal responses, and the functions of the genes that control these processes. We next explain how immune responses that are directed against apparently harmless molecules such as components of food, or the body itself, can cause serious disease (Section 7.3). Using case studies to provide examples we then outline what we know, and often still do not know, about the pathogenesis or genetic basis of some of these conditions, what we have learnt from animal models and sometimes how these conditions can be treated (Section 7.4). We then describe how the immune system can cause the rejection of foreign transplants and how it is that some types of transplant can directly attack the recipient (Section 7.5). We end by providing a very brief overview of the potential role of the immune system in helping to defend against not just infectious agents, but perhaps also tumour cells, and how these might fail and lead to cancer (Section 7.6).
7.1.1 Immunity, Disease and Therapy
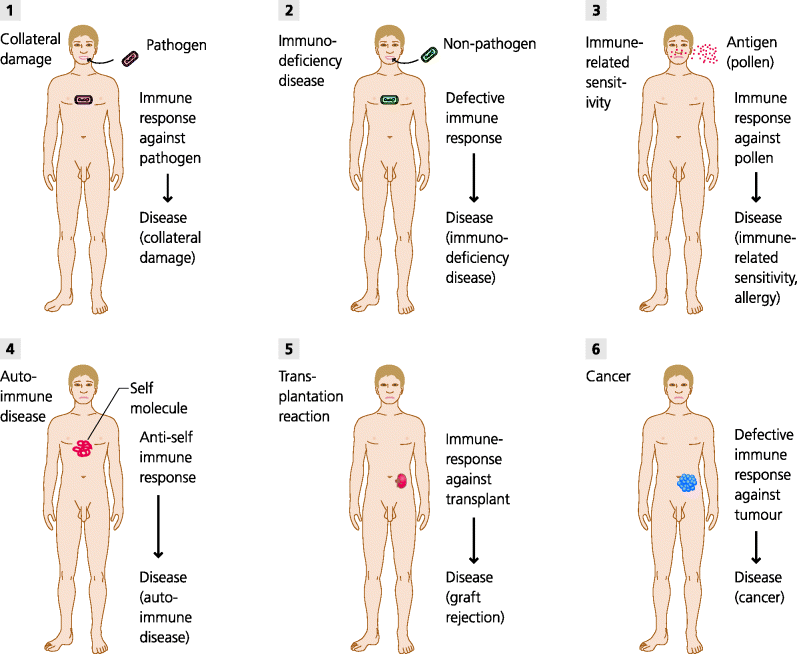
What are the diseases and conditions that relate to the immune system, and what forms of therapy are being developed to deal with them? See Figure 7.1.
Fig. 7.1 Immune-related diseases. Immune responses are necessary to eliminate pathogens. (1) These responses may, however, cause collateral damage to the host. (2) Defects in immunity can result in severe infections by agents that are not normally pathogenic; such immunodeficiency diseases can be primary genetic defects or secondary in cause (e.g. because human immunodeficiency virus (HIV) damages crucial components of immunite reactions). (3) Some individuals make responses against normally harmless substances (peanuts, pollen, fungal spores) resulting in allergies or other immune-related sensitivitie. (4) Other individuals may make immune responses against self components leading to autoimmune diseases. (5) Immune responses can also cause transplant reactions including rejection. (6) If, however, immune responses are not able to reject abnormal tumour cells, a cancer may develop. (Autoinflammatory diseases that result from abnormalities of the innate immune system are not shown, Section 4.2.2.4.)
7.1.1.1 Immunity and Disease: Immunopathology
Immunodeficiency diseases are conditions resulting from genetic or acquired defects where there is a failure of immune responses. By definition, immunodeficiency diseases result in severe, persistent, unusual or recurrent (SPUR) infections, and the type of infection is usually characteristic of the deficiency in question. These are introduced in Section 1.6.3.1, and are used as evidence for the importance of different cells and molecules in host defence against infection throughout other chapters of this book. Hence, we will not discuss them in detail in this chapter. Likewise other types of genetic defect that lead to dysregulation of immunity and lead to autoimmune and/or lymphoproliferative diseases, such as APECED (Autoimmune Polyendocrinopathy–Candidiasis–Ectodermal Dystrophy), IPEX (Immune dysregulation, Polyendocrinopathy, Enteropathy X-linked) and CTLA-4 deficiency, are discussed in 5.5.5.1 and not considered further in this chapter.
Immunopathology
In this chapter we focus initially on aberrant responses against self or harmless antigens that cause immune-related diseases. Collectively these are generally known as immunopathological conditions; they can also result from an aberrant or unwanted adaptive immune response to an infectious agent. The diseases we will consider mostly are mainly allergies and autoimmune diseases.
- Allergies, and other immune-related sensitivities, are sometimes termed hypersensitivity diseases. Well-known examples are hay fever, peanut allergy and, contact sensitivity to metals. These conditions result from an abnormal or exaggerated response to non-infectious, external antigens that are normally harmless to other individuals. Collectively we will refer to these foreign antigens that originate from our environment as extrinsic antigens.
- Autoimmune diseases. Well-known examples of these are rheumatoid arthritis, insulin-dependent (Type I) diabetes and multiple sclerosis. These represent the consequences of an abnormal or exaggerated response against non-infectious, internal antigens to which most individuals do not respond. Collectively we will refer to these self antigens that are part of the body itself as intrinsic antigens. Autoimmune diseases are commonly divided into organ-specific and systemic diseases. In the former case they involve one main organ in the body, such as the thyroid gland. In the latter case they affect several organs or body systems, such as the blood vessels and joints in systemic lupus erythematosus (SLE) and rheumatoid arthritis, respectively. An autoimmune disease may be precipitated by an extrinsic trigger, such as a viral or bacterial infection, but this is quite distinct from the intrinsic mechanisms that are involved in the persistence of disease once they have been induced.
In the last part of this chapter we discuss immunity in relation to transplantation and cancer.
- Transplant and transfusion reactions: Transplant rejection represents entirely normal, but unwanted, responses against foreign antigens on transplanted tissues (e.g. kidneys) or cells (e.g. bone marrow) that are derived from other people or different species. Such reactions lead to rejection of the transplant and are triggered by foreign antigens that are, in themselves harmless. Collectively these are called transplantation antigens. Conversely, bone marrow transplants can -host cause damage to the host; graft-versus-host disease. Blood transfusion is a special case of transplantation. Responses to non-self antigens such as the ABO or Rhesus blood group antigens expressed on erythrocytes lead to transfusion reactions.
- Tumour immunity: New, uncontrolled clonal growth of single cells and development into tumours may be associated with expression of new proteins by the cell. It is possible that the immune system could recognize these new antigens. Such an immune reaction might destroy or control these cells in the same way as infectious agents are dealt with this is called tumour immunosurveillance. Collectively these types of altered or aberrant self antigens are termed tumour antigens and, if they stimulate an immune response that eliminates the mutant cells, they can be termed tumour rejection antigens (by comparison with transplant rejection). In some cases, such as a virally induced tumour, tumour antigens may actually originate from infectious agents. However, in the case of clinical malignancy this form of rejection has clearly not worked, but it might be possible to stimulate the immune system to bring about rejection of the tumour.
A final, general category of immune-related diseases are the autoinflammatory diseases, where the conditions are caused by abnormal responses of the innate immune system and the adaptive system is not involved. These diseases result from genetic defects, but differ from primary immunodeficiencies (above) because these mutations frequently lead to a gain rather than a loss of function and do not lead to SPUR infections; these conditions are discussed in Section 4.2.2.4 and are not consider further in this chapter.
It is very important to bear in mind, throughout this chapter, that any immune response may involve inflammation, though not all immune responses do provoke inflammation per se. Also, some immune deficiencies result in inflammation either with or without infection (e.g. the autoinflammatory diseases). It is important to understand which diseases do include inflammation when considering different types of therapies for them.
7.1.1.2 Disease and Therapy: Immunotherapy
Immunotherapy
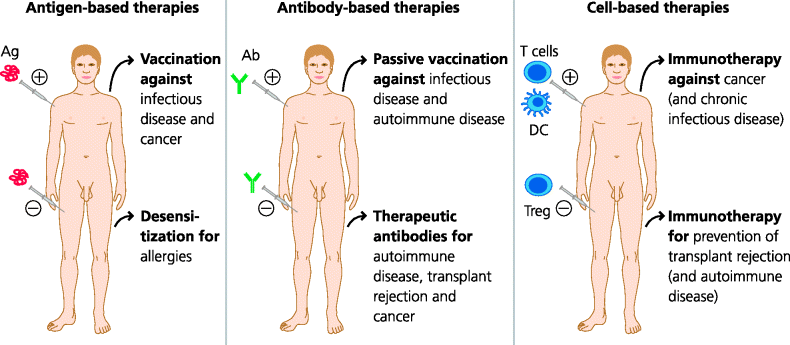
Throughout this chapter we will provide examples of the use of biological components of the immune system to treat ongoing disease, which may or may not have an immunological causation. We will not discuss vaccination in the prevention of disease since, this is covered in Sections 2.5 and 4.6. We will include antigen- and antibody-based therapies, therapy involving adoptive transfer of immune cells, and mention cytokine therapy. We will also mention some pharmacological approaches to therapy including inhibitory or immunosuppressive drugs. See Figure 7.2.
Fig. 7.2 Immune-related therapies. Antigen-based vaccines. A variety of agents ranging from DNA to attenuated living microbes can be used to induce a specific adaptive immune response that protects against subsequent infection. In the future it may be possible to vaccinate against cancers. Antigens can sometimes be used to reduce specific immune responses (e.g. desensitization in allergies). Therapeutic antibodies. Monoclonal antibodies that bind to and inhibit a molecule involved in a disease can be used to treat the disease. Such therapeutic antibodies are now used in some autoimmune diseases and cancers and in prevention of organ transplant rejection. Cell-based therapies. In the treatment of cancer, antigen-specific lymphocytes can be transferred or dendritic cells (DCs) used to induce specific responses. In the future it may be possible to use regulatory T cells (Treg) to suppress unwanted responses (e.g. in transplantation or autoimmune diseases).
- Antigen-based therapies: One of the best known examples of an antigen-based therapy is vaccination (e.g. as used to stimulate protective immunity against an infectious diseases; Section 2.5). In the context of this chapter, however, we will focus on the use of an antigen, or relevant epitopes from the antigen, to decrease or alter the immune response to that antigen. This can be done by administering the antigen in such a way that it will induce tolerance antigen unresponsiveness), often by triggering suppressive mechanisms mediated by regulatory (Treg) T cells or even inhibitory antibodies, or will re-programme the response in a non-harmful direction. To date this approach has been used mainly in the treatment of allergies. For example, desensitization to allergens involves injecting increasing doses of antigenic material, starting with very small doses.
- Antibody-based therapies: Pre-formed antibodies can be used in a variety of ways. For example, they can be used to deplete or inhibit cell populations expressing specific molecules or to block the activity of potentially harmful molecules such as cytokines. Particular human antibodies can be used to prevent certain conditions. However, more recently, monoclonal antibodies are being used in a variety of therapeutic settings. These therapeutic antibodies are being used to treat autoimmune diseases, to inhibit transplant rejection, and as therapies for certain cancers. In other cases, autoimmune diseases can be treated by depleting antibodies that cause damage (by plasmapheresis) while in others, paradoxically, they can actually be treated by giving large doses of pooled antibodies from normal donors, intravenous immunoglobulin (IVIG) therapy.
- Adoptive cell-based therapies: Key cells of immunity can be transferred to stimulate desired immune responses, or to inhibit damaging or unwanted immune responses. At present these forms of treatment are being explored mainly for the treatment of tumours and chronic infections, but may in the future be used for autoimmune diseases and transplantation. For example, dendritic cells (DCs), expressing the antigen of choice, may be injected in order to stimulate or modulate ongoing responses. Alternatively, tumour-specific T cells recovered from a patient can be expanded in vitro and re-injected into the same patient. Experimentally, Treg have also been used to suppress transplant rejection.
- Immunosuppressive drugs: This is a huge area and we will not cover it in depth in this chapter. Many pharmacological drugs are anti-inflammatory and are widely used to prevent transplant rejection or to control autoimmune diseases. Others interfere with key intracellular signalling pathways in cells such as T cells and are also widely used in these settings.
- Cytokine therapy: Given the potency and selectivity of cytokines, it is not surprising that their use in therapy has been widely explored, primarily in cancer and chronic infections such as hepatitis B and C, and tuberculosis. Type I interferons (IFNs) and various other cytokines (e.g. IL-2) have been trialled in different conditions, but the results to date are not encouraging due to the multiple biological effects these cytokines can stimulate and their sometimes serious side effects. Growth factors, (e.g. granulocyte-macrophage colony-stimulating factor (GM-CSF)), can be used, sometimes successfully, to mobilize cells (e.g. neutrophils) from the bone marrow in cases where the numbers of these cells are reduced.
In some cases, some of the above therapies are needed to treat the side effects of other therapeutic procedures that are themselves immune-related. Examples are the use of therapeutic antibodies to overcome rejection following organ transplantation, or to treat graft versus host disease (GVHD) following bone marrow transplantation.
7.2 What are the Mechanisms of Tissue Damage Caused by the Immune System?
7.2.1 Initiation and Effector Phases of Disease
There is nothing novel or unusual about the effector mechanisms that cause cell or tissue damage during an immune response. They are the same mechanisms used to combat infections and may, for example, involve antibodies, cytotoxic T cells or activated macrophages. It is just that in these pathological conditions, these the responses are inappropriate or unwanted. The great bulk of these diseases are initiated by inappropriate activation of CD4 T cells responses, which of course necessarily also involves the innate immune system. In this section we will concentrate on the effector mechanisms that cause the damage and disease. This area is relatively well-understood. In Section 7.3 we will then approach the difficult question of why such unwanted responses are initiated.
7.2.2 Classification of Immunopathological Mechanisms
In the 1960s, Gell and Coombs introduced a classification of immune-related diseases that is still in use today. They called these hypersensitivity reactions and divided them into four types (I–IV). However, we now know that many diseases usually involve more than one mechanism. It is crucially important, at the outset, to appreciate the key points:
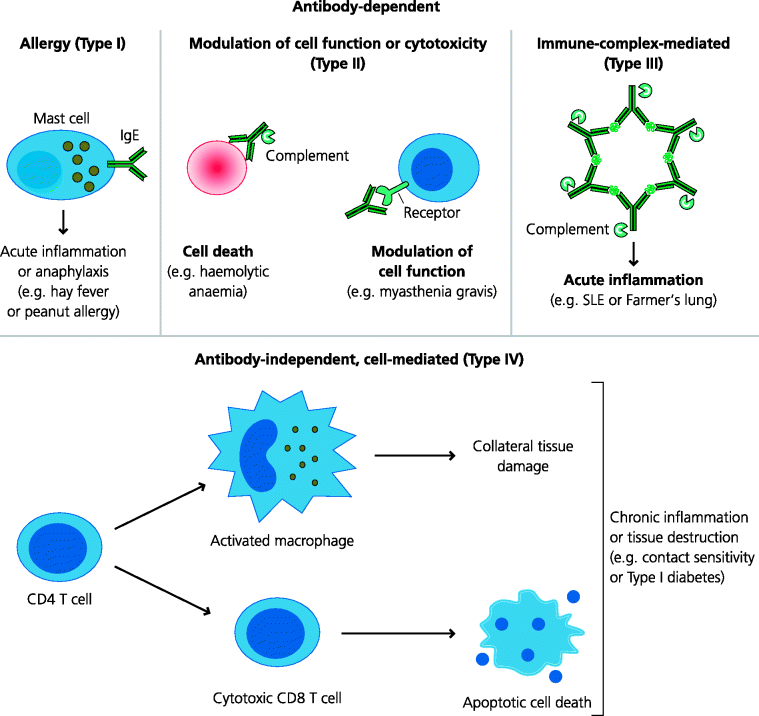
The first three types of hypersensitivity diseases depend on antibodies as the effector mechanism. Type I, allergic hypersensitivity or, simply, allergy, depends on the interactions of antigen with IgE that is bound to mast cells. Type II, originally termed cytotoxic hypersensitivity, depends on the ability of antibodies, often IgM or IgG, to kill cells or to modulate their functions. Type III, immune complex-mediated hypersensitivity, depends on the ability of antigen–antibody complexes to initiate inflammation. In contrast, Type IV, delayed-type hypersensitivity (DTH), is mediated by cellular responses such as those including CD8 T cells or activated macrophages that damage the tissue, and is independent of antibodies. Of course we now know that all these mechanisms are dependent, at least in part, on the inappropriate activation of CD4 T cells as the initiating immune event. However, at the time, Gell and Coombs could only classify these types according to the effector mechanisms they could identify. Hence, even though we have gained an increasingly sophisticated understanding of the different mechanisms that can cause tissue damage, and increasingly find that we cannot explain any given disease on the basis of a single mechanism alone, this classification remains in use today. See Figure 7.3.
Fig. 7.3 Mechanisms of immunopathology. Tissue damage caused by adaptive immune responses can be due to or independent of antibodies. IgE antibodies (IgE) cause allergy and anaphylaxis by acting through mast cells. IgG and IgM can modulate cell function directly, by killing the cell or by blocking or stimulating receptors. Immune complexes can cause acute inflammation locally or systemically. Cell-mediated (antibody-independent) immunopathology is usually due to Th1-biased responses involving cytotoxic CD8 T cells and/or activated macrophages, but in some cases Th2-biased responses are important such as in some forms of asthma.
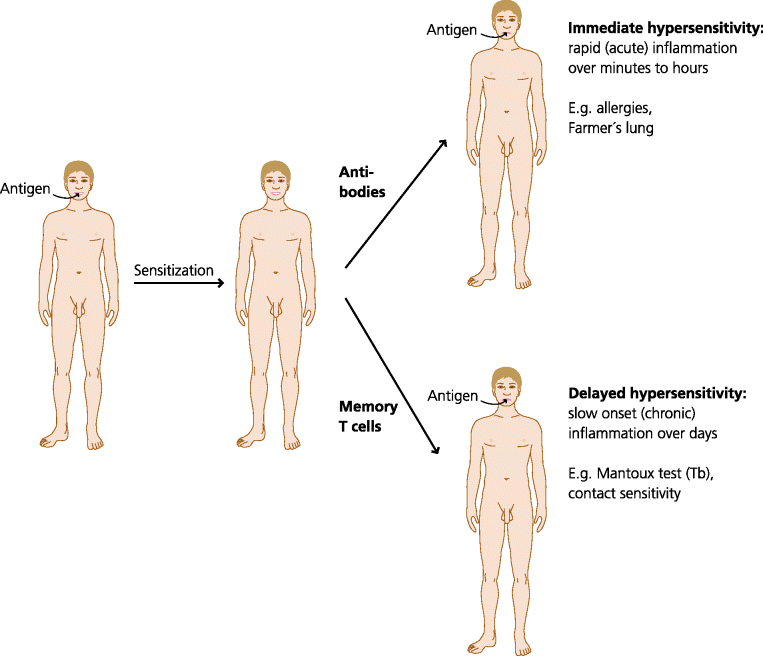
The types of immune response seen in hypersensitivity reactions are often divided into immediate and delayed responses. This refers simply to the time taken for a response to occur after antigen encounter in a sensitized individual i.e. one who has previously met the antigen and made a response. Immediate responses take only minutes to a few hours to happen because they represent the activity of pre-formed antibodies. In contrast, delayed responses usually take 24–48 h to appear. This is because they depend on re-activation of expanded populations of previously primed T cells, and their recruitment from the secondary lymphoid tissue and migration to the site where the antigen is present; this takes a relatively long time. Once again we stress that these responses are only seen in individuals previously sensitized to an antigen. When an antigen is encountered for the first time there is usually no overt response, although of course if the antigen hangs around for long enough, or is produced by a pathogen resident in the body, effector mechanisms can be activated and a reaction may be seen. See Figure 7.4.
Fig. 7.4 Immediate and delayed-type hypersensitivity reactions. If an individual who has been sensitized to a particular antigen is challenged with the same antigen, an inflammatory response may occur very rapidly, within minutes to hours. This is called immediate hypersensitivity, and is seen in allergies and conditions such as Farmer’s lung. Immediate hypersensitivity occurs, and can only occur, because there is pre-formed antibody. Other responses take much longer (24–48 h) to occur. This is called delayed hypersensitivity, and is seen in the Mantoux test for immunity to tuberculosis and contact sensitivity to substances such as nickel. We call these responses delayed due to time taken to reactivate and expand memory cells, but the response in fact accelerated relative to the primary T cell response that occurs during sensitization.
7.2.3 Diseases Caused by IgE Antibodies (Type I: Allergic Hypersensitivity)
IgE is normally synthesized in response to parasitic infections. True parasites can be divided into single-celled protozoa and complex, multi-cellular metazoa such as intestinal worms. IgE is generally synthesized only against the metazoa. In these situations IgE may have a protective effect by helping with worm expulsion, although given the very chronic nature of such infections, it is often clearly not effective.
Allergy, which is wholly IgE-dependent, is thought to be the only form of hypersensitivity reaction that does not have a counterpart in autoimmune diseases. Recently, however, it has been found that some SLE patients do have IgE antibodies to nuclear antigens, although it is not clear if these antibodies play a role in pathogenesis. Allergies are common; hay fever is probably the commonest immune disorder in the Western world. It can be a minor inconvenience (runny eyes and sneezing because of local inflammation) or fatal (death within minutes due to severe, systemic responses). We understand the mechanisms underlying allergy very clearly, but we have very little idea why some of us become allergic and others do not, even when they are in the same family or even identical twins; this is discussed below. Allergic reactions happen very quickly in sensitized individuals, typically within minutes of antigen exposure.
What does IgE do in allergy? The effector mechanisms are well understood. During the sensitization phase B cells are, for unknown reasons, switched to express IgE by activated CD4 T cells (Th2 cells Section 1.4.5.1). IgE binds to high-affinity IgE Fc receptors (FcRs) on mast cells in the absence of antigen. All of us who are allergic have mast cells coated with IgE specific for the particular antigen(s) to which we are sensitive. In this specific case the antigen(s) is referred to as an allergen(s). If a sensitized individual is re-exposed to that antigen, the antigen can bind to IgE coated mast cells at the mucosal surface or in the tissues. See Figure 7.5.
Fig. 7.5 IgE in defence and disease (type I hypersensitivity reactions). IgE is thought to give some protection against parasitic helminth worms. However, some individuals exposed to antigens such as grass pollen or peanuts make adaptive responses in which B cells switch to IgE production. The secreted IgE binds to high-affinity Fc receptors (FcRs) on mast cells. If the individual is re-exposed to the antigen, it can cross-link the IgE on mast cells. This induces the mast cells to release inflammatory mediators. Locally these cause acute inflammation but if they are released into the blood they may cause potentially fatal anaphylaxis. IgE responses are typically controlled by Th2 cells.
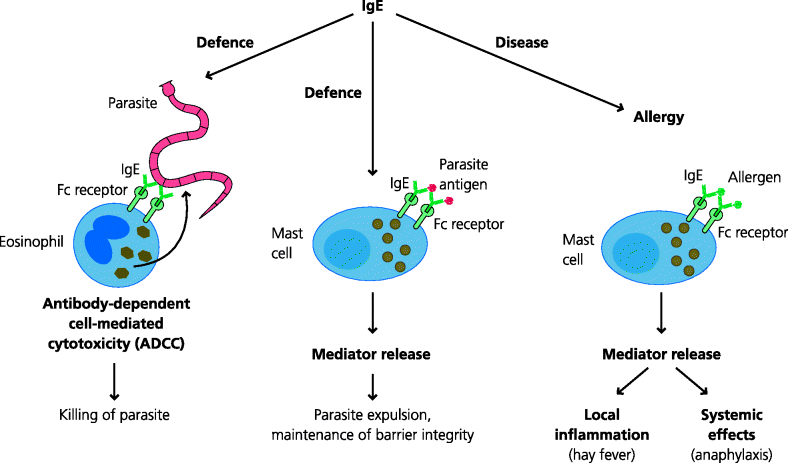
If the antigen is able to cross-link two or more IgE molecules, a signal is generated by the IgE FcRs to which they are bound. This results in the very rapid activation of the mast cell, within seconds to minutes. Mast cells have large cytoplasmic granules which are storage organelles for pre-formed mediators of inflammation. Amongst the latter one of the most important is histamine. Activation of the mast cells results in the contents of the granules being released into the extracellular spaces. Locally, histamine causes vasodilatation (e.g. red eyes), increased vascular permeability leading to swelling or oedema (e.g. blocked nose) and the stimulation of sensory nerves (e.g. itching). This is not usually serious, but think what will happen if you are sensitive to wasp stings and are stung in the back of the throat: not a good site for oedema as the swelling will block air entry to the lungs. It is even worse if the effects are body-wide (systemic). Histamine can cause widespread dilatation of veins, leading to a potentially catastrophic fall in blood pressure, and constriction of bronchial smooth muscle, causing respiratory obstruction. Together these features are called anaphylaxis and this is often fatal if not treated with adrenaline to reverse these effects. Mast cells additionally store pre-formed pro-inflammatory cytokines that are also released very rapidly after activation. After this very rapid response, the mast cells later synthesize other mediators such as the lipid metabolites prostaglandins and leukotrienes that also contribute to the oedema and bronchial constriction. Treatment of less severe reactions includes anti-histamines and non-steroidal anti-inflammatory drugs (NSAIDs) to inhibit synthesis of the lipid mediators.
7.2.4 Diseases Caused by Antibody-Dependent Modulation of Function (Type II: Cytotoxic Hypersensitivity)
In defence against infection. Antibodies act in several different ways: they can block binding of a pathogen or toxin to its target cell (neutralization), they can kill some pathogens or the cells they have infected through complement mediated lysis or antibody-dependent cell-mediated cytotoxicity (ADCC), or they can opsonize pathogens, leading to their phagocytosis and killing by cells such as neutrophils. All these mechanism have counterparts in some autoimmune diseases. The diseases caused by antibodies targeting host cells were called cytotoxic Type II hypersensitivity disease by Gell and Coombs, although in many cases we now know that cells are not actually killed. Importantly, in this type of mechanism, the antibodies are specific for molecules expressed on a cell surface, and are mostly IgM or IgG. See Figure 7.6.
Fig. 7.6 IgM and IgG in defence and disease (type hypersensitivity reactions). Antibodies are crucial in defence against many bacterial and viral infections, largely by opsonizing the microbe, or by neutralization, preventing binding of toxins, viruses or bacteria to cells. Occasionally lysis of bacteria may occur. In disease, antibodies can act on cells to kill them (as in haemolytic anaemia) or modulate their functions. For example, in myasthenia gravis, autoantibodies to the acetylcholine receptor block binding of acetylcholine and cause muscle weakness. IgG responses can be controlled by either Th1 or Th2 responses.
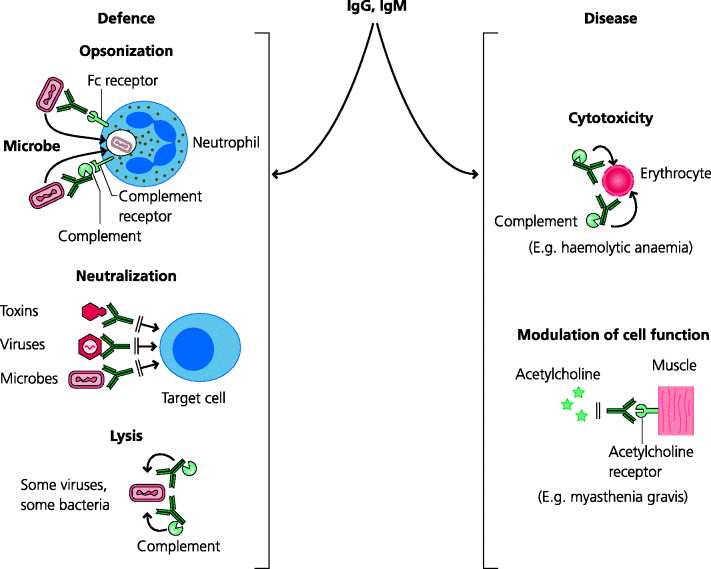
The most direct way of modulating cell function is by killing the cells. Cells that have bound antibodies can be killed in two ways. One is mediated by complement as in haemolytic anaemia and the other by a variety of cells (ADCC) (Section 6.2.6.4). Other than killing cells, antibodies can inhibit cell function by blocking functional molecules on cell surfaces. Thus, in myasthenia gravis, antagonist antibodies against the acetylcholine receptor makes muscle cells unable to respond to acetylcholine, leading to muscle weakness. In some cases (e.g. hyperthyroidism) agonist antibodies can cause abnormal stimulation of the receptor.
7.2.5 Diseases Mediated by Immune Complexes (Type III Hypersensitivity)
Although strictly, any antigen that is bound to an antibody may be called an immune complex, the term is usually used to describe multi-molecular complexes of antibodies and soluble antigens. Such complexes may form during any antibody response to a soluble antigen and are usually disposed of rapidly following binding of complement, which facilitates their uptake by macrophages. In some cases immune complex formation can be beneficial since they can enhance and regulate adaptive immune responses. For example, immune complexes with or without bound complement components, may be taken up by DCs, leading to increased T cell responses, or may bind to receptors on B cells and regulate antibody responses. In some cases, however, large, insoluble complexes are formed, which cannot be cleared by macrophages. These are potentially harmful and lead to Type III hypersensitivity reactions. In these reactions immune complexes act together with complement and/or neutrophils to stimulate acute inflammation, leading to tissue damage. See Figure 7.7.
Fig. 7.7 Immune complexes in defence and disease (type III hypersensitivity reactions). Immune complexes of soluble antigens and IgM or IgG antibodies bind complement and are normally cleared by macrophages after transport to liver and spleen by erythrocytes. Such complexes may also regulate immune responses following binding to DCs or B cells via FcRs or complement receptors. Large complexes cannot be cleared this way and are deposited locally in tissues or in small blood vessels in organs such as the kidney. Here, activated complement initiates acute inflammation and recruited neutrophils cause local tissue damage.
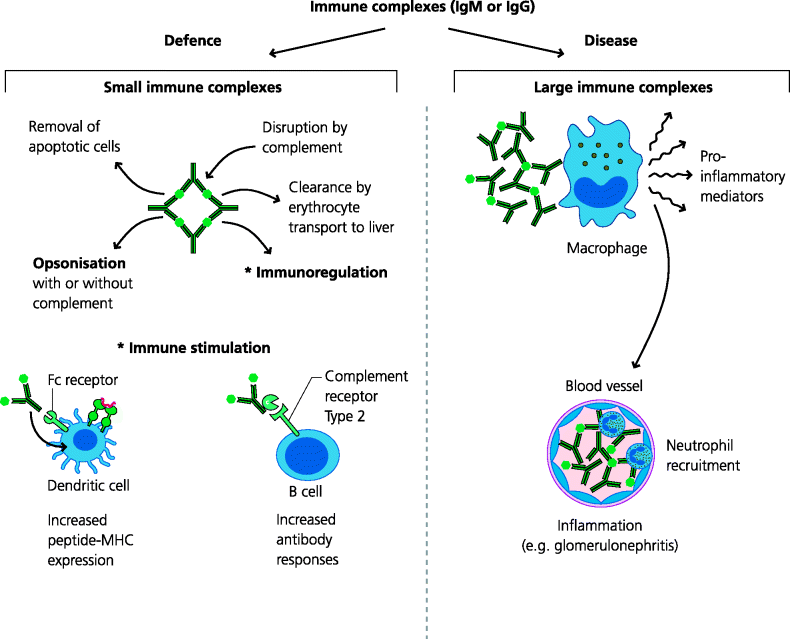
7.2.5.1 Diseases Caused by Blood-Borne Immune Complexes
Normally, circulating immune complexes bind to red blood cells (RBCs) via a complement receptor (CR1) and are transported to the liver and spleen where macrophages line blood vessels. Here the complexes are removed from the red cells, endocytosed and destroyed, allowing the red cells that transported them to continue to circulate. If, however, complexes form that cannot be removed, they remain in the circulation and are likely to be deposited in small blood vessels where, over time they cause disease. This may occur anywhere in the body, and these diseases are therefore called systemic, but the kidneys and joints are often particularly affected. Damaged tissues may also be the sites of deposition. For example, in SLE, complexes may be deposited in UV-damaged skin, leading to the characteristic facial butterfly rash. Wherever they are deposited, immune complexes will inevitably induce acute inflammation at the site of deposition. It is the inflammation that causes the disease. Inflammation results from the activation of complement and recruitment of neutrophils. Neutrophils bind the complexes via FcRs and complement receptors but may not phagocytose them, leading to the release of damaging enzymes and other molecules.
7.2.5.2 Disease Caused by Local Deposition of Immune Complexes
In the above examples the antigen–antibody complexes causing disease were systemic (i.e. the complexes were present in the blood). In some cases, however, complexes may be formed in situ at sites where antigens are deposited (e.g. in the lung following inhalation of antigens). As with systemic immune complex disease, the pathology is caused by acute inflammation and the symptoms reflect this local inflammation.
7.2.6 Diseases Caused by Cell-Mediated Immune Responses (Type IV: Delayed-Type Hypersensitivity (DTH)
Activation of CD4 T cells, particularly along the Th1 pathway, leads to the generation of potent effector mechanisms, (Section 1.2.2) crucial for recovery from infection, including cytotoxic CD8 T cells and activated macrophages. These mechanisms are also all potentially mediators of tissue damage. Collateral damage may occur in an infection if the pathogen is not effectively controlled, as in hepatitis B infection or tuberculosis (Section 2.4.1.1). Damage can also occur if the response to a harmless extrinsic or intrinsic antigen is not regulated, as in Crohn’s disease, a chronic inflammation of the intestines. The damage can be direct, for instance if cytotoxic CD8 T cells kill host cells expressing viral or intrinsic antigens. It can also be indirect, for instance if a macrophage secretes enzymes that can break down connective tissue (presumed to be one of the mechanisms of damage in tuberculosis and rheumatoid arthritis). The actual site of the damage may also be crucial to disease severity. Inflammation in the meninges of the brain may be fatal, whereas in other sites the same degree of inflammation can be relatively harmless. We now understand that many of the features of DTH responses involve aberrant activation of Th1 or Th17 cells (it is only more recently that Th17 cells were discovered and it seems that some responses previously thought to be due to Th1 cells are really caused by Th17 cells, Section 1.4.5.1). However, in some cases, Type IV sensitivities may involve aberrant activation of Th2 cells (e.g. in conditions where eosinophils accumulate in tissues). Hence, we can now discriminate between different types of DTH depending on the cells involved, but they are all thought to be largely or totally independent of See Figure 7.8.
Fig. 7.8 T cell-mediated immunity in defence and disease (type IV hypersensitivity reactions). Th1-biased CD4 T cells are essential for protection against many microbes via the activation of CD8 T cells or macrophages. If, however, CD4 T cells have been sensitized against a self or harmless foreign antigen, they can stimulate tissue damage, again via CD8 T cells or macrophages. Th2-biased T cells may be important in defence against some parasites, but are also responsible for the damage seen in conditions such as asthma.
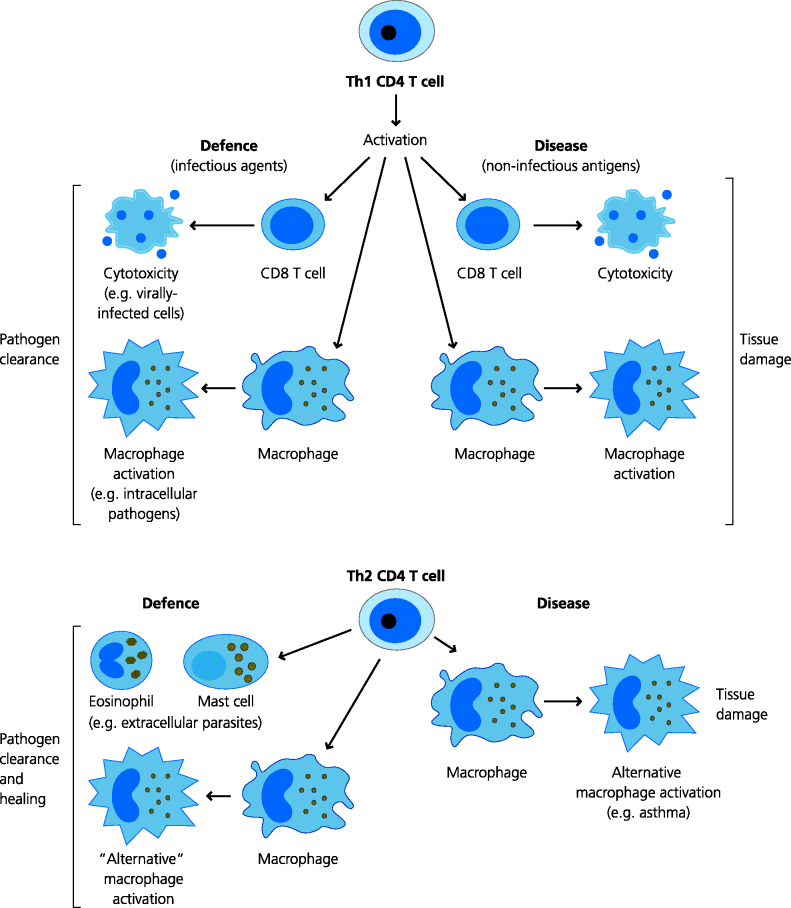
However, in many immunopathological conditions, both B and T cells are activated against extrinsic or intrinsic antigens. Thus, in a patient suffering from a type IV DTH disease, cytotoxic T cells, activated macrophages and antibodies may all be present. It is, however, very important is to know which of these effector mechanism(s) is or are playing a significant role in causing the tissue damage and disease since such information will inform the application and development of therapies. It can be difficult to determine the relative importance of different effector mechanism(s) in human diseases, but in animal models adoptive transfer of antibodies or cells can give clear indications. These aspects are discussed in later sections of this chapter.
7.3 Why Do We Make Harmful Immune Responses to Harmless Antigens?
In this section we will discuss why it is that immune responses can be made to harmless antigens that do not derive from pathogens, including extrinsic foreign and intrinsic self antigens, as well as transplantation and blood group antigens. We will first consider the antigenic repertoire of lymphocytes – how conventional T cell receptors (TCRs) and B cell receptors (BCRs) are generated, and how they can recognize antigens. We will then discuss lymphocyte activation, and why and how it may be that lymphocytes can be activated inappropriately, causing disease. This section will also discuss the polarization of immune responses in relation to immune-mediated diseases. Finally, we will introduce the genetic bases of some of these diseases and the roles that environmental factors may play in their pathogenesis.
7.3.1 Antigen Recognition by Lymphocytes in Normal and Pathogenic States
7.3.1.1 Generation of the Antigenic Repertoire of Lymphocytes
In this section, and this chapter in general, we focus almost exclusively on responses of conventional lymphocytes of humans and mice, which have highly diversified antigen receptors. These are the αβ T cells and the follicular B cells. Generally speaking, the potential roles of other types of lymphoid cells have been much less considered, and certainly much less studied, in most of the areas we will be discussing here. See Figure 7.9.
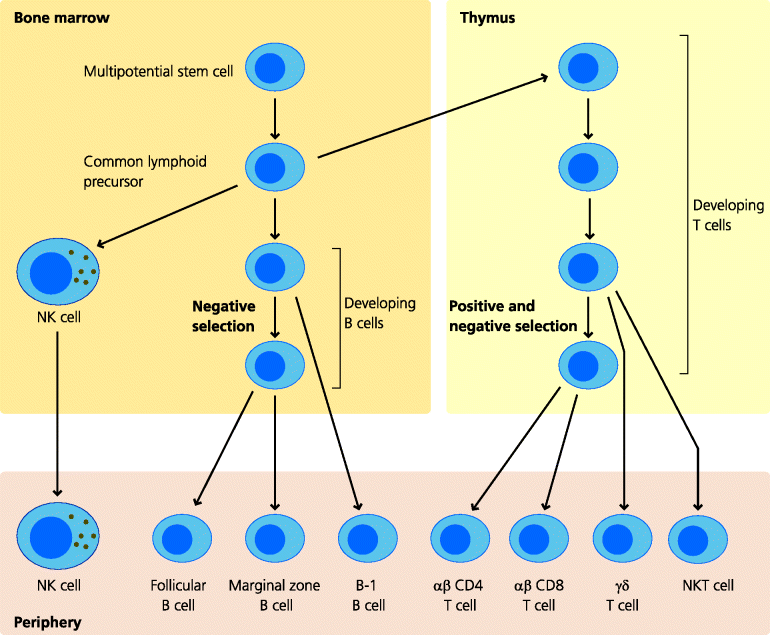
Fig. 7.9 Lymphocyte development. The multi-potential stem cell gives rise to the common lymphocyte precursor. This cell can develop into NK cells, B, T and NKT cells. NK cells and B cells complete their development in the marrow, while T and NKT cells complete theirs in the thymus. Conventional follicular B cells and αβ T cells undergo negative selection to reduce the chance of autoreactive cells being produced, although this process is incomplete (Figure 7.11). αβ T cells also undergo positive selection to ensure that their receptors are MHC-restricted. The interested reader might wish to consider whether or not, and if so to what extent, positive and negative selection might apply to the other cell types shown.
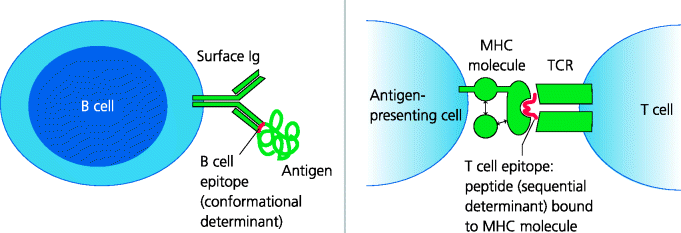
T cell activation, particularly of CD4 T cells, is crucial to almost all forms of immunopathology, including those where it is antibodies that cause the damage. Why can T cells potentially recognize self and normally-harmless foreign antigens? To understand this we need to re-consider the antigenic repertoires of lymphocytes. These repertoires represent the collective specificities of all the TCRs and BCRs that are expressed in an individual. The repertoire defines what can and cannot be recognized in the external (foreign) and internal (self) worlds. This means the range of antigens, or more precisely, the parts of antigens that can bind to the individual’s antigen receptors. In terms of antibody-mediated immunity these are termed B cell epitopes, more or less anything that is the right size and shape to fit into the binding site of a BCR. For conventional T cells, the antigens are usually peptide–major histocompatibility complex (MHC) complexes, which are often called T cell epitopes. See Figure 7.10.
Fig. 7.10 B cell and T cell epitopes. Conventional BCRs (surface Ig) and secreted antibodies recognize regions that are exposed on the surface of macromolecules as well as small molecules. The regions recognized are termed B cell epitopes or conformational epitopes. If an epitope is to be recognized it must have a relatively rigid three-dimensional structure. Conventional T cells, in contrast, recognize MHC molecules with bound peptides that have been produced by degradation of proteins. These peptides (that can be bound by MHC molecules and recognized by TCRs) are called T cell epitopes or sequential epitopes since the peptide is a linear sequence from the intact protein. In contrast, conformational epitopes for B cells can be formed by amino acids that are widely separated in the primary sequence of a protein.
TCRs and BCRs are generated by random rearrangement of gene segments and by other mechanisms that further increase diversity (Section 1.5.3.2). As a result of this randomness, the antigenic specificities of the receptors generated cannot be predetermined. Hence, there are specific mechanisms that are designed to ensure that these receptors function in the correct way and do as little damage as possible. However, while these are highly sophisticated mechanisms that work well in the population as a whole, they may be less than perfect at the level of individuals with immune-related disease.
In the case of T cells the peripheral repertoire is shaped by MHC molecules. This is because the randomly generated TCRs are selected so that T cells can only recognize peptides bound to self MHC molecules and not peptides alone. Hence, during their development in the thymus, MHC molecules present self peptides to the developing T cells. Only those developing T cells that weakly recognize a self peptide–self MHC molecule complex are allowed to develop further, while the remainder die within the thymus. This process is called positive selection. What this means is that the T cell repertoire is inherently self-reactive, although normally the strength of recognition of self peptide–MHC complexes by T cells is very weak (i.e. it is of low avidity).
Later in their development, T cells that recognize self peptide–self MHC complexes very strongly (high avidity) undergo apoptosis, so that highly autoreactive T cells do not escape the thymus. This process is called negative selection (Section 5.5.3). These selection processes ensure that those T cells that leave the thymus are generally not highly reactive against self peptide–MHC complexes expressed in the thymus. T cells are thus MHC-restricted, but non-reactive to many self peptides. Because it happens in the thymus, this process is called central tolerance. Negative selection is, however, necessarily incomplete, because it is limited by the range of self peptides that are expressed in the thymus. As it is not possible for all self peptides to be expressed in the thymus, self-reactive (autoreactive) T cells inevitably escape into the periphery. Negative selection of developing B cells also occurs, but this process turns out to be less stringent than for T cells. Hence, it is likely that even more potentially autoreactive B cells exist. It is therefore important to appreciate that self-reactivity of lymphocytes is a normal state in adaptive immunity and that the presence of these autoreactive cells does not necessarily cause us problems. Clearly, however, if such lymphocytes are activated, it is easy to understand how they could cause autoimmune diseases; without them, there could be no autoimmune diseases.
7.3.1.2 Promiscuity of Antigen Recognition by Lymphocytes
There is, however, another important consideration in relation to lymphocyte antigen recognition. TCR and BCR recognition is highly discriminatory since both types of receptor can recognize extremely small changes in their respective epitopes. For this reason recognition is sometimes said to be highly specific. However, this term is misleading. TCR and BCR antigen recognition is actually highly cross-reactive and very promiscuous. For B cells there is abundant evidence from structural and functional studies that any given antibody can bind with greater or lesser strength to a wide variety of different epitopes. We know that TCR recognition must also be cross-reactive because of the phenomenon of alloreactivity (Section 1.6.5.1). Astonishingly, it is estimated that maybe 1–3% of all the T cells in any individual can recognize a single MHC molecule from any other (allogeneic) individual (Figure 1.36 and Section 7.5.1.2, 7.5.5.1). Given that there are hundreds of different alleles of different MHC molecules in the population, and that this of alloreactive T cells frequency remains largely the same however it is tested, there must be a vast amount of overlap that represents an enormous promiscuity in TCR recognition. The only satisfactory explanation for alloreactivity is that it represents a very high frequency of T cells that have been selected to recognize viral or microbial peptides bound to self MHC molecules but which happen to cross-react with foreign MHC molecules, with or without their bound peptides.
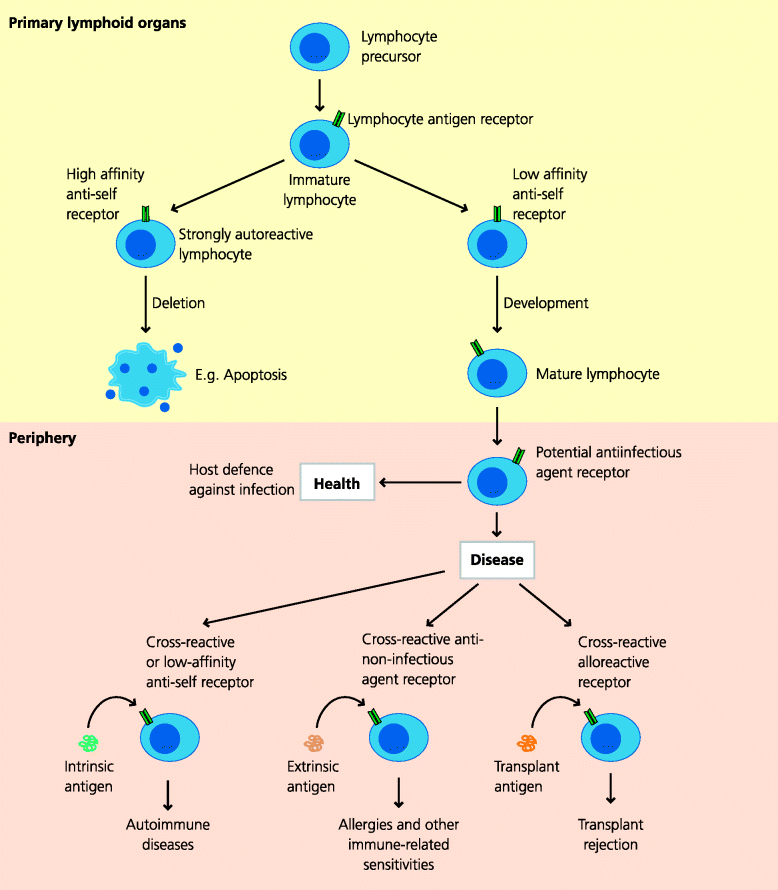
So the promiscuity of lymphocyte antigen recognition readily explains why allografts are rejected, and the extent of cross-reactivity in this setting explains why such immune responses are so vigorous. Cross-reactivity also explains why B cells can potentially recognize extrinsic antigens from the external world and make antibodies against food proteins or pollens, for example. Taking this one step further, it may also explain why such a response to an extrinsic antigen might also cross-react with an intrinsic antigen and cause collateral (autoimmune) disease. The same considerations apply to T cells in relation to peptide–MHC recognition. Hence, it is clear that lymphocytes must inevitably be able to recognize harmless antigens. However, to make any sort of response they must first be activated. Thus, the next question to be considered is why they are activated in these pathological settings. See Figure 7.11.
Fig. 7.11 Lymphocytes in health and disease. B and T cells as they develop express a pre-formed, anticipatory repertoire of antigen receptors that can potentially recognize antigens from infectious agents. For both B and T cells, cells that express receptors for self antigens that are present in their environment during development may be eliminated before they enter the periphery, including secondary lymphoid organs. This process is inevitably incomplete and some potentially self-reactive lymphocytes will enter the periphery where, under abnormal circumstances, they may induce autoimmune responses. Lymphocytes that express receptors for harmless self antigens such as pollen or food cannot be eliminated during development and unless peripheral mechanisms work to prevent their activation may induce immune-related sensitivities. A high frequency of T cells can also react against molecules expressed by other (allogeneic) individuals and can be responsible for transplantation reactions (rejection and GVHD).
7.3.2 Lymphocyte Activation in Immunological Diseases
Understanding lymphocyte activation in immunopathology is crucial for understanding the pathogenesis of the respective diseases, and for designing appropriate and effective therapies.
7.3.2.1 Signals for Lymphocyte Activation
Lymphocyte activation is essential for any adaptive response against an infectious agent. It is also essential for the development of any of the immunopathological conditions described above. We re-emphasize that all such pathogenesis probably depends on CD4 T cell activation. Activation requires that two different sets of signals are delivered to the T cell, and its subsequent fate (e.g. what type of function it acquires) is controlled by additional signals.
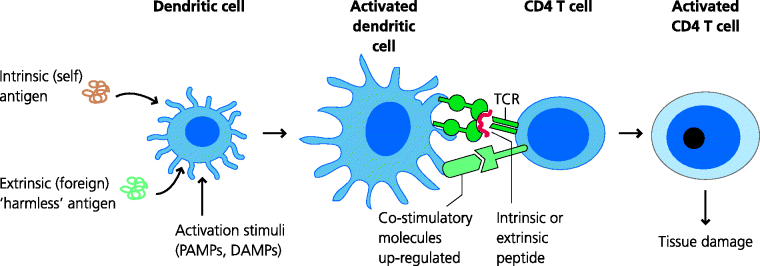
If a TCR recognizes a peptide-MHC with sufficient strength this generates a signal (Signal 1) that is essential for activation. Whether or not a T cell that has received Signal 1 does become activated is regulated by signals from costimulatory molecules, collectively known as Signal 2. Costimulation can be stimulatory (positive) or inhibitory (negative). The generation of costimulatory signals is regulated by signals from the innate immune system in response to the sensing of danger, such as that presented by infection. For example, DCs typically increase their expression of costimulatory molecules in response to pathogen-associated molecular patterns (PAMPs). These are conserved features of pathogens that are recognized by pattern recognition receptors (PRRs) such as Toll-like receptors (TLRs) (Section 1.2.3.1). Hence, they become able to trigger activation of T cells that recognize epitopes from the same pathogens. As another example, activation of complement and deposition of specific complement components on immune complexes, or potentially on surfaces, can also enhance the ability of B cells to make specific antibody responses. See Figure 7.12.
Fig. 7.12 CD4 T cell activation in immunopathology. Normally, DCs express intrinsic (self) or extrinsic (harmless foreign) peptides on their MHC molecules. If these DCs are not fully activated, when if they encounter a T cell specific for these peptide-MHC complexes the T cell will not be activated and may undergo apoptosis. If, however, the same DC has been stimulated by PAMPs or possibly DAMPs in the periphery, it will increase expression of costimulatory molecules and may become be able to activate the T cell, which will then have the potential of mounting a damaging response against the self or harmless antigen.
If a CD4 T cell does become activated it can then differentiate to express a particular set of properties, such as its capacity to secrete a selected group of cytokines or migrate to particular tissues. Which properties it does acquire is again determined by external signals coming primarily from innate immune and other non-immune cells; these can be loosely termed Signal 3. In immunity to infection, this control of T cell activation and differentiation is crucial as T cells, particularly CD4 T cells, determine which effector mechanisms are activated because different pathogens require different mechanisms for their elimination.
7.3.2.2 Lymphocyte Activation in Disease
We have seen that, inevitably some peripheral lymphocytes are able to recognize self or harmless foreign antigens. For these antigens, if potentially reactive lymphocytes do not meet their cognate antigen, or in the case of follicular B cells do not receive help from a T cell, harmful responses cannot occur. Even if T cells do recognize such antigens, under normal circumstances these potentially reactive T cells, and B cells, are controlled by regulatory mechanisms outside the thymus; we know this from the consequences of human defects in the genes for FoxP3 and Fas or Fas ligand, for example (Section 5.5.5.1). It is clear, however, that in some diseases these lymphocytes do become activated. Since we produce self-antigens continuously as part of cell renewal, are continually exposed to extrinsic harmless antigens, and as most of us carry persistent viruses, positive control mechanisms (regulation) are important in suppressing autoreactive responses. So how does the body normally avoid harmful reactions? Let us consider this in relation to the two signals for T cell activation (above).
Understanding Signal 1 is relatively straightforward. In immunopathological settings this, for example, requires B cell recognition of epitopes from extrinsic, intrinsic or transplantation (and possibly tumour) antigens, and T cell recognition of MHC-bound peptides derived from the same antigens. Understanding Signal 2 is more complex. In immunity to infection the pathogen expresses PAMPs that stimulate the innate system. In addition, it has been suggested that cell or tissue damage, whether or not this is associated with infection, generates damage- (or danger-) associated molecular patterns (DAMPS; Section 4.2.2.4) that, for example, can also lead to increased expression of costimulatory molecules by DCs, or perhaps activate complement. What, however, stimulates Signal 2 in responses to self or harmless foreign proteins? Self or harmless foreign antigens are not usually, by themselves, associated with PAMPs or DAMPs and would be predicted to induce active or passive forms of tolerance. See Figure 7.13.
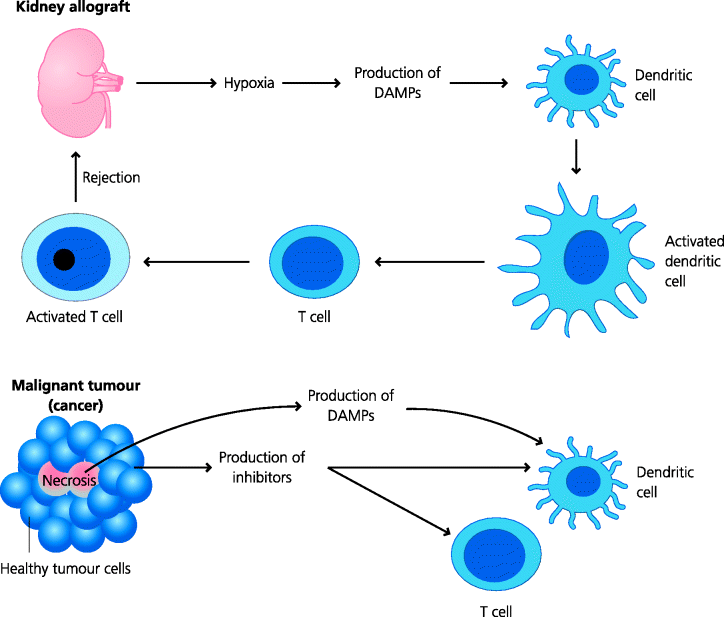
Fig. 7.13 Damage-associated molecular patterns. The initiation of adaptive immune responses to non-microbial antigens (e.g. allograft rejection) may depend on the release of molecules from dead or damaged (e.g. hypoxic) cells that activate innate immune cells such as DCs. These molecules have been termed DAMPs. Some candidate DAMPs such as uric acid and ATP have been identified. Malignant tumours often possess areas of necrosis that could release DAMPs; however, such tumours have evolved multiple ways of preventing adaptive responses from rejecting the tumour.
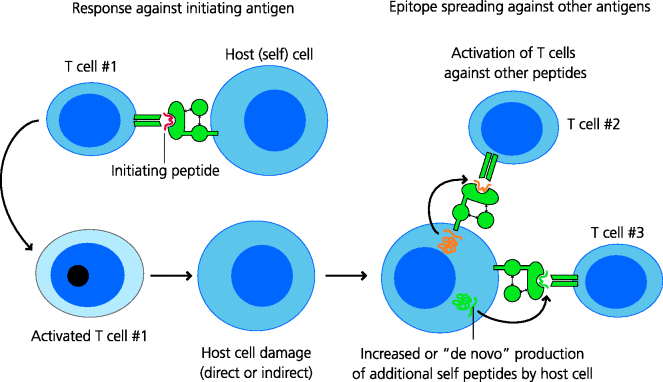
Frankly, in most autoimmune diseases we do not yet know why the immune response is triggered. In some there is clear evidence that infection is a trigger and it has been suggested that most or all autoimmune diseases may be precipitated by infections. Hence, PAMPs may induce costimulation that, with direct or cross-reactive recognition of the self antigen by autoreactive TCRs, may lead to T cell activation and the initiation of damage to the tissue. In such settings it is clear that the autoimmune disease is caused by direct recognition of an autoantigen. However, there is a further complication that can seriously hamper our understanding of which antigens are the most important in any given disease. This is because the initial damage to any tissue may then lead to exposure of self antigens to which T cells have not been tolerized (and potentially the generation of DAMPs). Hence, this can lead to the induction of new T cell responses to these antigens. This is termed epitope spreading. Epitope spreading is also seen in pathological B cell responses. See Figure 7.14.
Fig. 7.14 Epitope spreading. In established autoimmune disease, T cells may be found that respond to a variety of antigens from the cells or tissues affected. This does not, however, mean that these antigens were involved in the initiation of the disease. A single peptide could be responsible for the initiation of the response, but subsequent damage to the cells, perhaps with DAMP involvement, means that other peptides may then be presented in an immunostimulatory form to T cells. This is known as epitope spreading and may serve to increase the overall strength of an autoimmune response.
In the case of other allergies and immune-related sensitivities (hypersensitivity diseases) again, in many instances, we do not know why these are triggered and why only some individuals exposed to these antigens develop the disease. Some allergens are proteases, which may modify self proteins or cause damage to tissues. In such cases, even though no infection has occurred, DAMPs may be induced or released to stimulate the induction of costimulation. Together with TCR recognition of the allergen, this may trigger an immune response that ultimately damages the tissue (although the sensitization phase may be silent). This is often the case in contact sensitivity, where a molecule applied to the skin triggers T cell activation, probably against self proteins that have been modified by the agent. In many cases, however, it is just not clear how the response is triggered.
Transplants represent another form of harmless antigen, yet grafts from different members of the same species (allografts) or from different species (xenografts) are rejected by vigorous responses. Any transplant is, however, subject to unnatural stimuli during its retrieval, including an inadequate supply of oxygen, leading to hypoxia for organ grafts, or storage at 4°C under non-physiological conditions for these and bone marrow grafts. Clinically, it has been observed that the shorter the time between removal of the organ from the donor and its transplantation, the more likely it is to be successful. In part this reflects the damage caused to the organ by hypoxia, but it also seems probable that this hypoxia induces the expression of DAMPs in the organ that induce innate immune activation, leading to increased costimulation.
Malignant tumour cells are antigenically very similar to the cells from which they arose. Why should we think that they might potentially induce an active response? Most tumours possess areas of necrosis, often caused by the tumour outstripping or invading its own blood supply, and this may stimulate the expression of DAMPs. The observation that many tumours have developed mechanisms to avoid the adaptive response is itself very good evidence that these tumours do have the potential to induce immunity, and that the immune response is selecting for mutants which can evade the response. Thus, tumour cells can evade T cell recognition by down-regulating MHC expression, and hence not generating Signal 1. In addition, they may produce molecules that inhibit expression of positive costimulatory molecules by DCs, and may also themselves express negative costimulatory molecules. Thus, they avoid generating, or actively modulate, Signal 2.
In immunity to infection, if the infectious agent is successfully eliminated, the immune response is no longer stimulated and a regulated set of negative costimulatory (and other) signals then helps to dampen down the response. If, however, the pathogen is not removed, ongoing immune stimulation can lead to serious collateral tissue damage (e.g. as may happen in tuberculosis). Similarly, in allergy and other immune-related sensitivities, if the antigen is removed the damage stops, for example in hay fever when the pollen season is over or in coeliac disease if gluten is avoided. In autoimmune diseases, however, there is a more or less unlimited source of antigen from self tissues and the immune response may continue indefinitely, as in rheumatoid arthritis, or until the tissue is completely destroyed as is the case for pancreatic β cells in Type I diabetes.
7.3.2.3 Polarization of CD4 T Cells in Immunopathology
CD4 T cell responses to infection show more or less polarization, at least in mice. This is an essential part of the response (being induced by Signal 3 in the case of T cells) and permits the generation of appropriate effector mechanisms (Section 1.4.5.1). Many autoimmune and other immunological diseases also show marked polarization of CD4 T cells. Type I (insulin-dependent) diabetes is an example in which marked Th1 polarization results in macrophage- and CD8 T cell-mediated damage. In contrast, the rare disease eosinophilic gastroenteritis shows marked Th2 polarization in terms of the antibody isotypes produced and the marked infiltration of eosinophils into the intestinal submucosa. In many, maybe even in all, cases, however, this polarization is not complete and any immune-related disease can involve a mix of mechanisms. For example, asthma can be viewed as a combined Th1 and Th2, and in some cases Th17, disease. In fact it is now suggested that in many diseases originally thought to result from Th1 polarization, the CD4 T cells are actually polarized towards Th17.
Polarization may reflect long-term stimulation of T cells in many immune-related diseases. In mice, polarization needs repeated antigenic stimulation, but we have relatively little understanding of the signals that induce these biases in humans. That the genotype of the individual plays a major role is clear from studies of autoimmune diseases in twins, but at present no genes have been identified which clearly explain polarization in disease (below). Understanding polarization does, however, hold promise for therapy. If a Th1-polarized disease such as Crohn’s disease could be redirected so that the activated T cells became polarized towards Th2, there would be a real possibility of therapy. In one trial in which Crohn’s disease patients were infected with a porcine intestinal worm (Trichuris suis, which induces a Th2 polarized response) a remarkably beneficial effect was observed. Similarly, allografts are generally acutely rejected by Th1-biased responses. If we knew how to redirect the response towards a Th2 type or how to induce Treg, it might be easier to prevent rejection. In the case of tumours, during their development the tumour microenvironment may induce Treg that suppress any otherwise beneficial responses, even if these could be induced. Again, understanding how inhibit or deplete these cells, or to re-programme the response, could lead to effective immunotherapy.
7.3.3 Genetic Basis of Immunopathology
Genes control immune responses in many different ways. We know much about this from analysis of immune responses to model protein antigens and from studies of susceptibility to infection. It is thus not surprising that pathological and unwanted immune responses are strongly influenced by genetic factors (e.g. the MHC in allograft rejection). In this section we will concentrate on autoimmune diseases and discuss other abnormal responses where there is relevant evidence. Whilst the effects of genes are easy to determine in inbred, homogeneous mice, it is much more difficult in out-bred humans. Studies in populations of consanguineous families, where gene sharing is common, have been very informative as have those in twins.
7.3.3.1 Concordance in Twin Studies
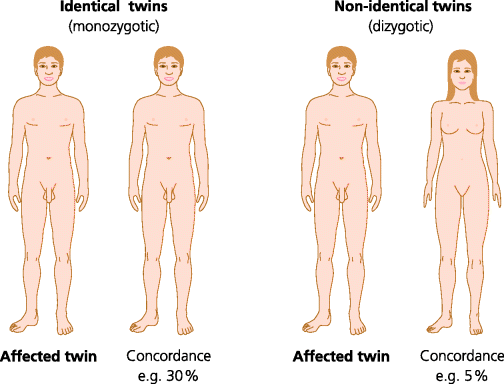
Studies of the incidence of disease in twins provide valuable evidence about the genetic basis of the disease. If one twin has a disease, the frequency with which the other twin also develops the disease can be estimated. This frequency is termed the concordance. If the concordance is higher in identical (monozygotic) twins than in non-identical (dizygotic) twins this is strong evidence for a genetic influence in susceptibility. In autoimmune diseases and immune-related sensitivities the concordance is frequently much higher in monozygotic twins. However, the degree of concordance in monozygotic twins is often in the range of 20–40%. (Coeliac disease resulting from sensitivity to oral gluten is an exception since the concordance in identical twins is 70–90%). The usual explanation for a low level of concordance is that environmental factors, such infections, play a major role in pathogenesis. See Figure 7.15.
Fig. 7.15 Disease concordance in twins. The genetic contribution to a disease can be estimated by studying sets of identical (monozygotic) and non-identical (dizygotic) twins where one twin has the disease. If the frequency of the disease in the other twin (concordance) is higher in identical than non-identical twins this is strong evidence for a genetic contribution. In most autoimmune diseases concordance is less than 50% in identical twins.
There is, however, another consideration to take into account in relation to the concordance between twins. Although we think that identical twins are wholly identical, this is only true of their genotypes. TCRs and BCRs are not identical in monozygotic twins. The combinatorial nature of receptor gene assembly means that a huge number of different receptors can in theory be generated. Any one individual will generate only a small subset of these possible re-arrangements and thus it is just not possible that identical twins will have identical sets of TCRs. If the possession of a particular TCR predisposes to an immune-related sensitivity or autoimmune disease, it is unlikely that both identical twins will express this particular TCR. It is not clear, however, whether these differences do play a part in explaining low-concordance.
Another possible reason for non-concordance relates to epigenetics. Gene expression in an individual cell can be modified in ways that can be inherited by daughter cells when the cell divides (e.g. by DNA methylation or histone modification). This is a new area, but studies suggest that epigenetic modification of immune-related genes can differ between monozygotic twins, and that these differences increase with age and geographical separation. The importance of epigenetic modification is, however, far from fully understood at present.
An important observation from human studies is that it is not uncommon for one individual to suffer from two or more autoimmune diseases concurrently. Thus, of individuals with ankylosing spondylitis (an arthritic disease of the spine), around 15% will have psoriasis (an inflammatory disease of the skin), 3% will have Crohn’s disease and 5% will have ulcerative colitis (inflammatory bowel disorders). Of patients suffering from asthma, 7–13% will have Crohn’s disease and 8–12% ulcerative colitis. These percentages are much higher than are seen in the general population, and strongly suggest that there are allelic variants of genes that bias those individuals who possess them towards the development of immune-related diseases.
7.3.3.2 Which Genes Confer Susceptibility or Resistance to Immunopathology?
Twin studies can identify a genetic basis for disease, but they cannot tell us which genes are involved. Identifying the actual genes involved is a major goal for clinical geneticists. Much information has come from linkage analysis. These are studies in which the association of disease susceptibility with other phenotypic traits can identify the region of a chromosome involved in susceptibility. Now that the human genome has been sequenced, candidate genes in this region can be sequenced and examined for differences (i.e. polymorphisms) that could explain susceptibility. More recently genome-wide association studies have been used. In these, the genomes of large numbers of individuals with and without the disease are scanned very rapidly using a very large number of genetic markers called single nucleotide polymorphisms (SNPs). This technique can enable us to determine if a particular gene(s) shows differences between affected and non-affected individuals.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


