Chapter Outline
T-Cell Receptor Subtypes and Specificity
Early T-Cell Commitment and Seeding
Stages of Intrathymic T-Cell Receptor αβ T-Cell Development and Spatial Considerations
T-Cell Recognition of Peptide-Major Histocompatibility Complex Complexes
T-Cell Receptor Rearrangement and Selection in the Thymus
Positive Selection, Negative Selection, and CD4/CD8 Lineage Commitment
B-CELL ACTIVATION AND FUNCTION
Thymus-Dependent Versus Thymus-Independent B-Cell Responses
B-Cell Receptor Triggering and Signaling
Coreceptors Involved in B-Cell Activation
Follicular B-Cell Differentiation: the Germinal Center
T-CELL ACTIVATION AND FUNCTION
T-Cell Receptor Triggering and Activation
Coreceptors Involved in T-Cell Activation and the Immune Synapse
Naïve T-Cell Differentiation to Effector and Memory T Cells
Helper T-Cell Differentiation and Function
Cytotoxic T-Cell Differentiation and Function
Control of the Immune Response: Regulation and Termination of T-Cell Activation
NONCONVENTIONAL T-CELL SUBTYPES
DEVELOPMENT AND FUNCTION OF T-Cell Receptor γδ T CELLS
NATURAL KILLER CELL DEVELOPMENT AND FUNCTION
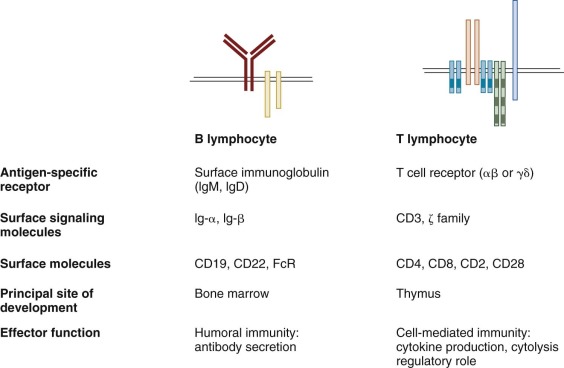
The immune system is a complex network of cells that serve to discriminate “self” from “nonself”—that is, to recognize one cell as one’s own and another cell as foreign or infected. This capacity helps protect the organism from infection by foreign pathogens (bacteria, viruses, and parasites) and from malignant degeneration. In addition, the cells involved in the immune response mediate rejection of foreign allografts. Although immune cells recognize and respond to all manner of foreign elements, at the same time they must not respond inappropriately to self-antigen. Education of immune cells results in a state of immunologic tolerance—that is, the absence of destructive antiself immune responses. Failure of self versus nonself discrimination, or failure of immunologic tolerance, may result in autoimmunity.
Whereas collectively a number of different kinds of cells are involved in the response to infection, the cells involved in mediating adaptive immunity are lymphocytes ( Table 23-1 ). The adaptive immune response is broadly classified into two categories: (1) humoral immunity, mediated by antibodies produced by B lymphocytes and their progeny; and (2) cell-mediated immunity, dependent on cell-cell interactions and mediated by T lymphocytes. Both T and B lymphocytes are derived from early hematopoietic stem cells (HSCs) and are distinguished not by morphology but by rearrangement of their antigen-specific receptors, by function, and by the expression of different cell surface molecules. In addition to adaptive immunity, natural or innate immune mechanisms also work to defend an individual from pathogens (see Table 23-1 ). Natural killer (NK) cells, also derived from bone marrow progenitors, serve as principal components of innate immunity and are discussed briefly later in this chapter. In addition, polymorphonuclear leukocytes (neutrophils), eosinophils, macrophages, and other myeloid cells are important components of the body’s early defense against microorganisms and are discussed in Chapter 22 .
| Innate Immunity | Acquired Immunity | |
|---|---|---|
| Specificity | Nonself | Random (in the context of self) |
| Effector cell type | Polymorphonuclear leukocytes (PMNs, neutrophils), natural killer cells, macrophages, eosinophils, etc. | Lymphocytes: B and T cells |
| Cell surface receptors | Germline | Rearranged |
| Distribution | Nonclonal | Clonal |
| Kinetics of the initial response | Rapid and immediate | Slow |
The acquired or adaptive immune response is mediated in large part by the recognition of a variety of cell surface proteins encoded by genes of the major histocompatibility complex (MHC). The MHC, located in the human genome on the short arm of chromosome 6, encodes two classes of proteins, MHC class I and MHC class II, that are highly polymorphic within the population and involved in the immune response. In humans these genes are termed human leukocyte antigens (HLA class I and HLA class II), as defined further later. In addition to HLA determinants, the MHC also encodes several secreted proteins of the complement system, selected immune modulators such as tumor necrosis factor (TNF), and specific proteins required for proper processing of antigen.
In addition to self-nonself discrimination, the adaptive immune response is further characterized by immunologic specificity and by memory. Acquired resistance to one antigen does not confer resistance to another unrelated (non–cross-reactive) antigen. The immune response must be capable of eliminating a foreign pathogen and developing mechanisms so that a second exposure to the same agent results not in worse disease but in more rapid elimination of the foreign agent. Cells previously exposed to and specific for one foreign protein or antigen respond more rapidly and more effectively to subsequent exposure to that same antigen, thereby leading to an anamnestic or secondary immune response. The immune response is highly regulated; cooperativity among different cellular elements and between cells and soluble mediators leads to modulation (i.e., amplification or suppression) of the immune response.
Technical advances, including single-cell cloning, monoclonal antibody production, generation of transgenic and knockout animals, and sequencing of the genome, have led to a veritable explosion in the number of cell surface proteins, cytokines, and chemokines discovered. The nomenclature has evolved and can be confusing. Cell surface proteins are often named by a cluster of differentiation (CD) assignment; the website http://www.uniprot.org/docs/cdlist contains links to protein information listed by CD name. Interleukins, the name given to soluble factors secreted by leukocytes or that act on leukocytes, or both, can be found along with their receptors at this site of the Human Gene Nomenclature Committee: http://www.genenames.org/genefamily/il.php . This chapter will focus on specific or acquired immunity and principally on the differentiation and function of B and T lymphocytes.
Lymphocyte Development
All B cells, T cells, and NK cells derive from pluripotent HSCs that arise first in fetal liver and then postnatally in the bone marrow. HSCs differentiate in the bone marrow and migrate to the appropriate lymphoid organs. These HSCs express cell surface CD34, a heavily glycosylated protein of 105 to 120 kd. Although CD34 serves as a marker for hematopoietic progenitors, its expression is not limited to these cells alone. CD34 is also expressed on early lymphoid (but not pluripotent) progenitors, endothelial cells in small vessels, some embryonic fibroblasts, and some cells in the nervous system. CD34 may be involved in cell-cell adhesion and may be important for inhibition of hematopoietic differentiation. CD34+ hematopoietic precursors give rise to both myeloid and lymphoid early progenitors. The traditional paradigm makes a strong distinction between HSC contained within the CD34+ compartment and lineage committed progenitors, which lack long-term reconstitution potential. Early myeloid differentiation derives from common myeloid progenitors (CMPs), which can give rise to common granulocyte-monocyte precursors (GMPs) or megakaryocyte-erythroid precursors (MEPs). The common lymphoid progenitor (CLP) differentiates into cells of the T (and NK) and B lineage. Dendritic cells can be generated in vitro from CD34+ bone marrow cells exposed to certain cytokines, but the relevant in vivo precursor population remains controversial, and CLP, CMP, and GMP are all shown to contain dendritic cell potential. New evidence from murine studies suggests that in single cell transplants, individual cells defined as HSC can give rise after a single division to one long-term repopulating cell and one myeloid committed cell ; thus lineage commitment may occur in individual cells at the HSC stage. How this translates to the human situation has yet to be uncovered. T-lymphocyte differentiation occurs largely in the thymus and is discussed in detail later. B-cell maturation occurs in the bone marrow and peripheral lymphoid organs.
Naïve B and T cells released into the peripheral blood have exquisite specificity. These resting B and T cells circulate in a quiescent state and function only after encountering cognate antigen (i.e., the specific substance that the lymphocyte recognizes). Recognition of antigen alone is generally insufficient to trigger lymphocyte activation; both B and T cells require other so-called costimulatory signals , in the form of cell surface receptor engagement or soluble factors, for effector function. Recognition of antigen by T cells and the costimulatory signals for both B and T cells are typically mediated by accessory cells of the innate immune system called antigen-presenting cells (APCs). Lymphocytes activated by specific antigen in the appropriate costimulatory context proliferate and mature into effector cells, a portion of which eventually differentiate into long-lived memory cells. The specifics of peripheral maturation, costimulation, and function of B and T cells are detailed in later sections.
Lymphocyte Specificity
Immunologic specificity is conferred by antigen-specific receptors expressed on the surface of B and T lymphocytes and by specific antibodies (immunoglobulins [Igs]) secreted by B cells and plasma cells ( Fig. 23-1 ). An individual lymphocyte has only one specific antigen to which it can respond. Each T cell expresses a unique T-cell receptor (TCR) and each B cell a unique Ig that can either be displayed on the surface (B-cell receptor [BCR]) or be secreted. The membrane BCR and secreted Ig of the B cell interact with epitopes expressed by native antigens, largely protein and nonprotein antigens such as complex epitopes of glycoproteins, lipids, and nucleic acids. Unlike Ig, the TCR recognizes small (most commonly 9 to 11 amino acid long) processed oligopeptides bound within the cleft of an MHC molecule on the surface of an APC. Dendritic cells, B cells, monocytes-macrophages, epithelial Langerhans cells, and (in humans) endothelial cells function efficiently as APCs. These APCs bear costimulatory molecules on their cell surface (discussed later), secrete cytokines, and process antigen; it is the complex formed between the processed peptide fragment of antigen and MHC proteins that interacts with the TCR. Exposure to antigen induces activation of only lymphocytes that bear Ig receptors or TCRs specific for that antigen. Any individual, however, has approximately 10 7 to 10 9 different T-cell and B-cell specificities. It is this clonal variability and diversity of the lymphocyte repertoire that maintain effective protection against foreign invasion.
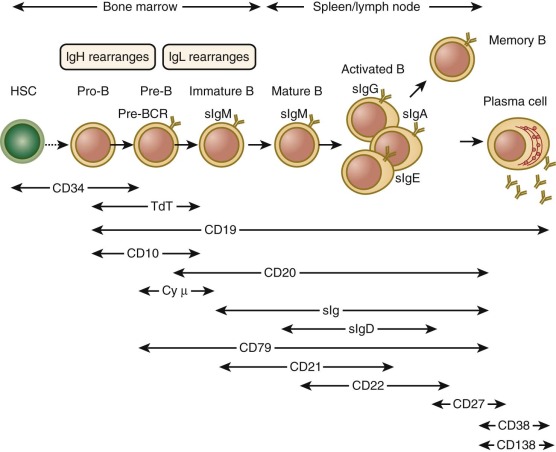
B-Cell Ontogeny
The first phase of B-cell maturation occurs in the bone marrow, where HSCs committed to B-lymphoid lineage develop into immature IgM+ IgD− B cells. The immature B cells exit the bone marrow, circulate in peripheral tissues, and mature further ( Fig. 23-2 ). The most critical steps in B-cell development at this stage involve the expression of Ig molecules on the surface as BCR, whose specificity is the same as the antibody that will ultimately be secreted by activated mature B-cell progeny. The diversity of the B-cell repertoire is due in large part to random rearrangement of multiple germline genes for Ig that are spliced to encode a unique Ig molecule for each cell. Highly regulated checkpoints ensure that only B cells that successfully rearrange Ig genes are selected for survival. Elimination of B cells whose receptors have high affinity for self-antigens is important to prevent autoreactivity. Finally, immature B cells released from the bone marrow complete their development in the spleen and differentiate into at least two distinct lineages of mature naïve B cells: follicular B cells and marginal-zone B cells.
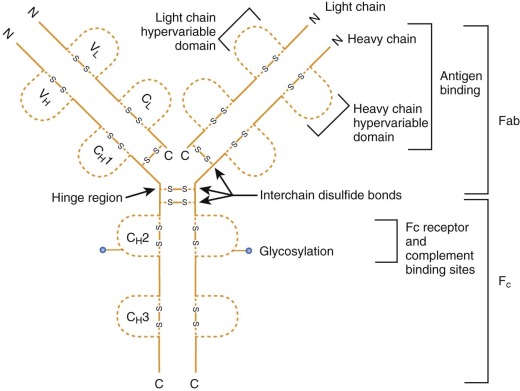
Immunoglobulin Structure
The structures of antibody molecules all share remarkable conservation of secondary conformation. The Ig-like fold is a domain shared among diverse proteins, each with signature hydrophobic common cores and common disulfide bridges. The antibody molecule is the prototype member of the Ig supergene family, a group that includes not only Ig but also TCR, the MHC, and a number of costimulatory molecules. Each Ig molecule is composed of two light chains of approximately 25 kd and two heavy chains of approximately 50 or 70 kd ( Fig. 23-3 ); each chain is composed of multiple Ig domains. B-cell antibodies can be subdivided by heavy-chain isotype and further subdivided by subclass ( Table 22-2 ).
There are five classes of antibodies—IgM, IgG, IgA, IgD, and IgE—defined by expression of the heavy chain: µ, γ, α, δ, and ε. The Ig-heavy chains contain sites for binding of Fc receptor (FcR) and complement, and each subtype appears to have a particular role in immune defense. The first class of antibody to be produced during development is IgM, which is expressed on the surface as a monomer and secreted as a pentamer. B cells that have undergone class switching (discussed later) express IgG, IgA, IgD, or IgE on the surface as the BCR or secrete a splice form of the Ig with the same heavy-chain isotype and specificity but lacking a transmembrane region (or both). The function of IgD in humans is not clear, although its expression is a marker for later B-cell development. IgA is secreted as a dimer and is found at mucosal surfaces such as the intestine. The bulk of circulating Ig is in the form of IgG, which is monomeric and further subdivided into four subclasses: IgG1, IgG2, IgG3, and IgG4. Persistently low serum levels of one or more IgG subclasses, in the presence or absence of IgA deficiency, may cause individuals to be susceptible to recurrent infections. Though controversial, prophylactic use of intravenous Ig has been suggested.
There are two subclasses of light chains: κ and λ (see Fig. 23-3 ). Both subclasses appear to serve similar functions. Monoclonal disorders of terminally differentiated mature B cells or plasma cells (e.g., multiple myeloma) express cells of a given light chain—that is, either κ or λ, but not both.
The structure of each Ig molecule is determined by hydrophobic interactions and disulfide bridges between the two heavy chains and two light chains (see Fig. 23-3 ). For each chain the N-terminal domain has a variable or polymorphic amino acid structure (V H for heavy chain, V L for light chain), whereas the C-terminal portions are constant within each heavy-chain or light-chain class (termed C H 1, C H 2, C H 3 for heavy chain and C L for light chain). These domains form pairs, V H with V L , C H 1 with C L , and C H 3 with C H 3. Proteolytic digestion of Ig by papain results in two Fab fragments, each composed of the V H /V L , C H 1/C L domains, and an Fc fragment composed of the two C H 2 and two C H 3 domains. The binding region of Ig that recognizes antigen is composed of portions of the V H and V L domains. Within each variable region there are three portions with increased amino acid variation, called hypervariable regions (HV1, HV2, HV3) or complementarity-determining regions (CDR1, CDR2, CDR3). The highest variability is found in CDR3, and all three CDRs from both chains participate in the binding of antigen. Variability within each variable region and each CDR and pairing of a unique heavy chain with a unique light chain all contribute to the combinatorial diversity of Ig specificity.
Immunoglobulin Rearrangement and B-Cell Maturation in Bone Marrow
The genes encoding the heavy chains of Ig are located on human chromosome 14, whereas the genes encoding κ and λ light chains are located on chromosomes 2 and 22, respectively. The heavy- and light-chain loci are each composed of variable (V), diversity (D), and joining (J) gene segments that recombine to yield the V H and V L domains, which in turn form the antigen contact surface of the antibody molecule and confer specificity. In addition, the heavy-chain locus contains nine heavy-chain constant (C) region genes (µ, δ, γ1, γ2, γ3, γ4, α1, α2, and ε constant regions) that define the mature Ig isotype.
Ig rearrangement occurs serially, and cells failing to rearrange the Ig genes successfully are eliminated and do not develop further ( Fig. 23-4 ). B-cell maturation also results in cell surface protein expression patterns that are used to define the pro–B-cell, pre–B-cell, and immature B-cell stages of development. CD34+ CD19− CD10− progenitors that have not yet committed to the B lineage have Ig genes in the germline configuration. The earliest B-cell precursors express the pan B-cell marker CD19 and the early marker CD10. They also express components of the rearrangement machinery such as recombination-activating genes (RAGs) and thus have early gene rearrangements detectable. Rearrangement begins with one heavy-chain allele; one of the 29 D H exons is spliced to one of the 6 J H exons, the intervening D and J sequences are eliminated, and a DJ segment is produced. The pro-B cell then splices one of the approximately 50 V H exons to the DJ segment to form a V(D)J segment. Finally, the µ constant region of the heavy chain (C µ ) is spliced onto the V(D)J segments.
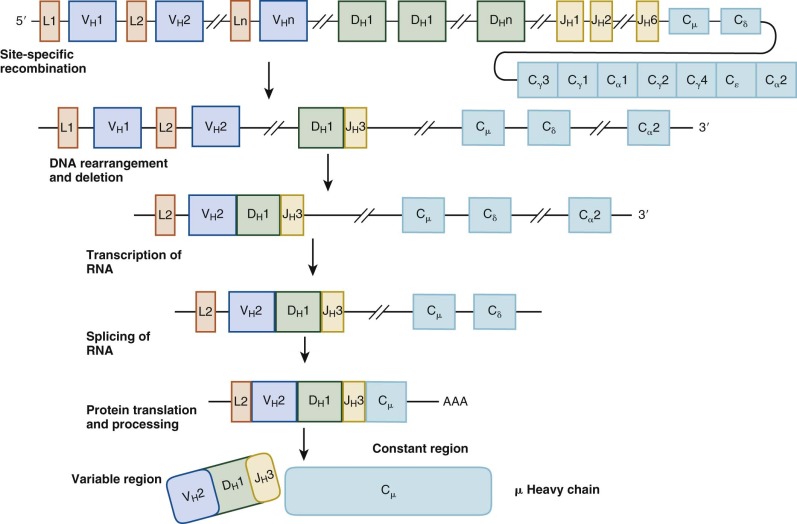
Differentiation to the pre-B stage is triggered by expression of intact µ heavy-chain protein or cytoplasmic µ, which occurs only in cells that bear in-frame productive V(D)J-C µ rearrangements. If rearrangement of the first heavy-chain locus is unsuccessful, the second allele undergoes rearrangement. Because the light-chain genes have not yet been expressed, cytoplasmic µ complexes instead with the nonpolymorphic surrogate light-chain proteins λ5 and VpreB. Cytoplasmic µ, surrogate light chain, and the accessory signaling molecules Ig-α (CD79a) and Ig-β (CD79b) together form the pre–B-cell receptor. Ensuing signals induce proliferation and instruct the cell to begin rearrangement of one of the κ light-chain loci. Pre-B cells lose CD34 expression, continue to express CD19 and CD10, and additionally upregulate CD20. The signaling cascade downstream of the pre–B-cell receptor is highly analogous to that of the mature BCR and activates a number of critical kinases, including Bruton tyrosine kinase (Btk) (discussed later). Boys born with mutations in Btk, manifested as X-linked agammaglobulinemia, have normal numbers of pro-B cells but lack pre-B cells and their progeny. Thus formation of the pre–B-cell receptor along with activation of downstream signaling is a critical checkpoint in this stage of maturation.
Light-chain genes rearrange by a process similar to the recombination of heavy-chain genes. The variable region of the κ gene, located on chromosome 2, is spliced to the J gene segment to produce a VJ region, which then splices to the one constant region (C κ ). If nonproductive, the second κ locus will rearrange. If both attempts to rearrange the κ locus are unsuccessful, the λ locus, located on chromosome 22, will begin to rearrange. Productive rearrangement of any light-chain locus prevents further B-cell rearrangement and also generates survival signals. Pre-B cells that successfully rearrange a light-chain gene will then express mature IgM protein, which appears as surface IgM (sIgM) complexed to Ig-α and Ig-β, thus forming the BCR. Immature B cells are sIgM+, CD79+, CD19+, CD10+, and CD20+, but they have not acquired IgD expression, which occurs after peripheral maturation.
Expression of the tissue-specific recombination-activating genes RAG-1 and RAG-2 is required for recombination. In addition, diversity is enhanced by the fact that the junctions between the D and J regions and the V and DJ regions are not precise; not only can the nucleotides vary, but one or more nucleotides (termed N regions ), not encoded in the genome, may also be randomly inserted (N-region diversification). The nuclear enzyme terminal deoxyribonucleotidyl transferase (TdT) is the polymerase that adds random nucleotides to recombination junctions and may have other roles. Recruitment of TdT to the junctions appears to depend on the Ku molecule (specifically Ku80), a heterodimer that binds DNA ends and is required for V(D)J recombination and DNA double-stranded break repair. Other proteins critical for nonhomologous end rejoining include Ku70, DNA-dependent protein kinase, DNA ligase IV, XRCC4, and the recently cloned protein Artemis. Defects in the recombination machinery in genes such as RAG1, RAG2, DNA ligase IV, and Artemis all result in severe combined immunodeficiency (see Chapter 24 ).
Peripheral B-Cell Maturation and Differentiation
Immature B cells must undergo further maturation in the spleen before acquiring the phenotype and functions of a naïve mature B cell. Naïve mature B cells are resting cells in G 0 phase; express the pan B-cell marker CD19, the marker CD20, and sIgM; and in contrast to immature bone marrow B cells, have lost CD10 expression and gained IgD expression. A similar population in the mouse is believed to be the common precursor for both follicular and marginal-zone B cells, named for their location within the spleen, discussed later.
The development of early B precursors into immature B cells is a process that requires Ig rearrangement and expression of the pre–B-cell receptor and BCR, but it does not require recognition of antigen. Maturation to the naïve resting B cell involves selection of B cells that recognize antigen (positive selection) while eliminating B cells whose BCR recognize self-antigens with high affinity (negative selection). It has been proposed, on the basis of murine data, that determination of mature B-cell fate relies on the relative signal strength and specificity of the BCR, with higher and lower signal strength driving to the follicular and marginal-zone lineages, respectively. Negative selection in particular is important for the deletion of potentially autoreactive clones and induction of B-cell tolerance. Indeed, it is estimated that 55% to 75% of immature B cells in humans recognize self-antigens, and thus a minority of developing B cells pass the negative-selection test.
T-Cell Ontogeny
T lymphocytes, like B lymphocytes and all other blood cells, are derived from self-renewing pluripotent HSCs. T cells are unique, however, in that their development also requires passage through a specific organ, the thymus. Indeed, the absolute requirement for the thymus is made clear by human congenital immunodeficiency states such as the DiGeorge syndrome, in which thymic organogenesis and therefore T-cell development are absent (see Chapter 24 ). Similar to B-cell ontogeny, the generation of millions of diverse T cells is accomplished by serial rearrangement of germline genes encoding the receptor for antigen recognition, the TCR. The bulk of peripheral T cells are of either the CD4+ helper or the CD8+ cytolytic lineages.
T-Cell Receptor Subtypes and Specificity
The TCR is a cell surface receptor capable of recognizing discrete antigens bound to the MHC or to MHC-related molecules expressed on the surface of APCs. TCRs are disulfide-linked heterodimers of either αβ or γδ proteins. TCR αβ−bearing T cells constitute the vast majority of circulating T cells, most of which recognize peptide antigens bound to polymorphic MHC class I or class II molecules. A subset of TCR αβ T cells, representing approximately 1% of all T cells in humans, in contrast recognize lipid antigens bound to the MHC class I–related nonpolymorphic molecule CD1 and are termed CD1-restricted or NK T cells, discussed in a later section. TCR γδ–bearing T cells account for a tiny fraction of the circulating T cells in humans but are very prevalent in intestinal tissue and are thought to recognize nonpeptide antigens (discussed later). The functions of CD1-restricted TCR αβ T cells and TCR γδ T cells are somewhat distinct from those of conventional TCR αβ T cells. The discussion of intrathymic T-cell development in a subsequent section will focus on the major conventional subset of T cells.
Early T-Cell Commitment and Seeding
The molecular events associated with and responsible for commitment to the T lineage have been increasingly elucidated in both murine and human models over the last 10 years. The question of whether T-lineage commitment occurs within the thymus or the bone marrow has been actively investigated. In addition to pluripotent CD34+ HSCs, a number of other more committed progenitors in bone marrow with specific cell surface marker characteristics have been identified with T-lymphoid potential, primarily in the mouse, but these progenitors in general also retain the capacity to differentiate into other lymphoid lineages, including the B and NK lineages. That various subsets identified in the human thymus similarly retain bilineage potential (i.e., NK/T potential or NK/dendritic cell potential) has led to the notion that the final steps in T-cell commitment occur in the thymus rather than the bone marrow.
In addition to losing the capacity to differentiate into non–T-lineage cells in vitro, T-lineage commitment is characterized by the upregulation of certain cell surface markers ( Fig. 23-5 ) and T-cell–specific genes important for the execution of downstream developmental programs. CD34+ cells in human thymus can be further subdivided by the expression of CD38 and the MHC-like molecule CD1a, with progression from the CD34+ CD38− CD1a− stage to the CD34+ CD38+ CD1a− stage to the CD34+ CD38+ CD1a+ stage. Acquisition of CD1a+ is largely associated with the loss of NK, dendritic cell, and plasmacytoid dendritic cell differentiation in vitro. Likewise, CD1a+ cells express the recombinase-activating gene products RAG1 and RAG2, and early TCR rearrangements are detectable after the acquisition of CD38 and CD1a. Transcription factors upregulated during these stages of early human T-cell commitment, whose requirement for T-cell development have also been demonstrated in gene-deficient mice, include GATA3 and Notch.
Interactions between thymocytes and epithelial cells are critical for events in later thymic development, such as positive and negative selection, as detailed later, as well as for early commitment and development. The so-called nude mouse , which lacks expression of the epithelial transcription factor Foxn1 , is born athymic, and humans lacking FOXN1 have severe T-cell deficiency, alopecia, and nail dystrophy. Unlike B cells, which can be induced in vitro to mature from precursors by co-culture on bone marrow stromal cells, T cells generally cannot be differentiated in vitro by this means, only when seeded into a three-dimensional source of primary thymic stroma, such as whole thymic organ culture. This distinction implies that specific soluble or cell-cell contact factors present only in the three-dimensional primary organ are required to engage developing thymocytes and induce their differentiation. The Notch signaling pathway, which is activated by the interaction of Notch ligands on epithelial cells and Notch family members on thymocytes, has emerged as one such factor. Expression of activated Notch1 in human HSCs is sufficient to induce T-cell development in vitro and in immunodeficient mice, and deletion of Notch1 from mouse thymocytes severely impairs early T-cell development. Interestingly, primary three-dimensional thymic stroma expresses Notch ligands such as Delta-like 1 and 4, and this expression is lost when the architecture is disrupted. Indeed, expression of Delta-like 1 or 4 in the OP9 bone marrow stromal cell line is capable of directing mouse and human uncommitted HSCs and even embryonic stem cells into the T lineage when co-cultured in a two-dimensional format in vitro. This powerful system underscores the importance of thymocyte-epithelial interactions during intrathymic development and raises the intriguing possibility of differentiating and expanding T cells in vitro for use in human therapeutics, which has been pursued in murine systems and recently in expansion of umbilical cord blood units to expand access to this source of therapeutic stem cells.
Stages of Intrathymic T-Cell Receptor αβ T-Cell Development and Spatial Considerations
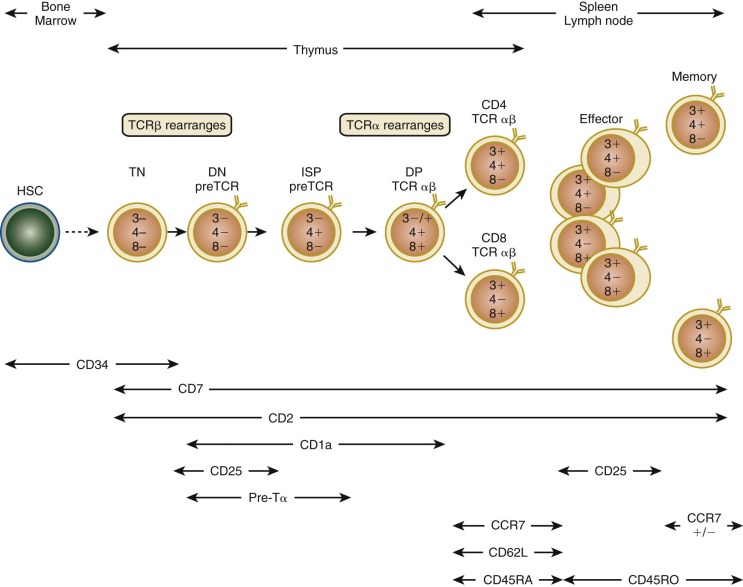
T-lymphoid precursors that enter the thymus from the blood mature through a series of ordered developmental stages characterized by changes in cell surface markers, sequential expression of the TCR genes, and predictable migration from the cortex to the medulla (see Fig. 23-5 ). The earliest thymocytes reside in the cortex, are CD34+, express pan-T-cell markers such as CD2 and CD7, but lack the TCR-associated αβ heterodimer and CD3-associated subunits of the TCR complex and the helper and cytotoxic mature T-cell coreceptors CD4 and CD8. Hence, these thymocytes are CD3− CD4−CD8−, or triple negative. During αβ T-cell development, triple-negative thymocytes that succeed in rearranging a functional TCRβ chain express TCRβ complexed to the invariant pre-TCRα protein. Pre-TCR signaling in thymocytes promotes rapid expansion and differentiation into double-positive (DP) thymocytes expressing both the CD4 and CD8 coreceptors and initiates rearrangement of the TCRα gene. In humans, in contrast to mice, there is an additional intermediate stage in which thymocytes express only CD4 and not CD8 (intermediate single positive [ISP]). DP thymocytes account for approximately 80% to 90% of the total thymocyte number and can be found in the cortex and at the corticomedullary junction. Although the majority of DP thymocytes die by apoptosis, those that successfully undergo positive and escape negative selection survive, commit to either the CD4/helper or CD8/cytotoxic lineage, and migrate to the thymic medulla before terminal maturation and export to the periphery.
T-Cell Recognition of Peptide-Major Histocompatibility Complex Complexes
A hallmark of the cellular immune response is antigen specificity, which is conferred by the rearranged and selected TCR on the surface of T lymphocytes. The TCR recognizes processed fragments of foreign proteins embedded in MHC molecules. There are two forms of MHC molecules: MHC class I (the major determinants are HLA-A, HLA-B, and HLA-C in humans) and MHC class II (the major determinants are HLA-DR, HLA-DQ, and HLA-DP in humans) ( Fig. 23-6 ). MHC class I molecules are composed of a 42-kd transmembrane, polymorphic α chain encoded in the MHC and noncovalently associated with a 12-kd soluble, nonpolymorphic (non-MHC) β chain termed β 2 -microglobulin. X-ray crystallographic analysis of HLA molecules in which peptide was embedded has demonstrated that the first two domains of the α chain both form α helices that together form a cleft ( Fig. 23-7 ). This cleft forms the binding domain for peptide antigen and accommodates a 9– to 11–amino acid fragment in an extended conformation; residues that interact with the MHC and those that interact with the TCR can be defined. Polymorphism within the MHC itself serves to ensure variation in the affinity of peptide binding; the extraordinary MHC polymorphism within the species further ensures that any single microbe is unlikely to mutate such that it is unable to bind all MHC molecules in the population and therefore escape T-cell recognition.
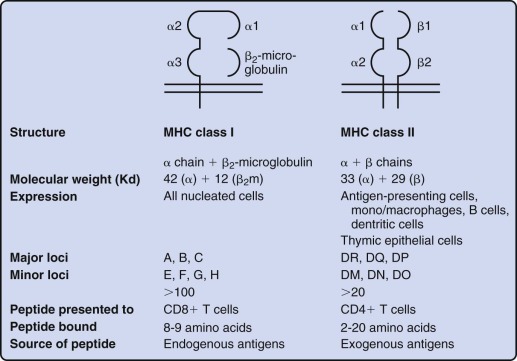
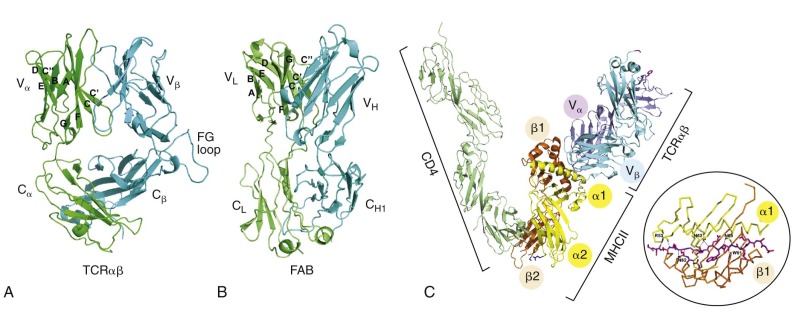
MHC class II molecules are formed by the noncovalent association of two transmembrane glycoproteins, αβ, both of which are polymorphic and encoded in the MHC. Solution of the structure of MHC class II molecules has shown that a peptide-binding cleft is formed by the first domains of each of the αβ chains; however, the ends of the cleft are open, unlike the situation in MHC class I molecules (see Fig. 23-7 ). The peptide-binding cleft of MHC class II accommodates processed peptides that are 10 to 30 (mean of 14) amino acids in length. As with MHC class I, the genetic polymorphism of MHC class II determines the affinity and specificity of peptide binding and T-cell recognition. Although MHC class I molecules are expressed constitutively on all human nucleated cells, MHC class II molecules are constitutively expressed only on B cells, monocyte-macrophages, and dendritic cells. Expression of MHC class II proteins may be induced on the surface of monocyte-macrophages, fibroblasts, endothelial cells, and certain mesenchymal and epidermal cells by a variety of inflammatory mediators and cytokines such as IFN-γ. In humans but not in mice, MHC class II proteins can be inducibly expressed on T lymphocytes as well, thus rendering T cells capable of antigen presentation to other T cells. These two classes of MHC proteins generally interact with different classes of T lymphocytes: CD4+ T cells recognize peptide antigen in association with MHC class II molecules, whereas CD8+ T cells recognize peptide antigen in association with MHC class I molecules. CD8 binds to invariant portions of the α3 domain of MHC class I and CD4 to a hydrophobic crevice between α2 and β2 domains (see Fig. 23-7 ).
A growing number of drug allergies have been reported and shown to be a consequence of their drug binding to MHC molecules. In general, peptides associate with HLA molecules by inserting parts of their amino acid residues into a set of six binding pockets in the HLA. The structure of these pockets is highly allele-specific, thereby dictating peptide-binding preferences for each HLA molecule. Remarkably, a recent study demonstrated that life-threatening drug-induced immune responses occur during abacavir treatment of HLA-B*57:01+ individuals as well as carbamazepine treatment of HLA-B*15:02+ patients through their binding to a specific pocket of the respective HLA molecules. Drug binding shifts the repertoire of self-peptides normally complexed with that HLA molecule for surface display. As a consequence, self-peptides never before arrayed on the surface of cells within the patient’s body are expressed and perceived by the T cell immune system as foreign. These led to the CTL-mediated destruction of such peptide arrayed target cells. It is likely that other drugs, environmental chemicals and toxins, and bacterial products function in a similar way, including interactions with class II MHC molecules. Given the immunologic basis of many cases of severe aplastic anemia (SAA), it is conceivable that the etiology of some or most SAA cases might be linked to such a mechanism.
T-Cell Receptor Rearrangement and Selection in the Thymus
Effective adaptive immunity relies on the random generation of a diverse repertoire of antigen-specific T cells, along with appropriate quality control measures to ensure that nonfunctional T cells are not needlessly allowed to mature fully. To achieve this goal, the TCR genes undergo somatic rearrangement in a serial fashion, and the growth and proliferation of developing T cells are tightly linked to signaling and transcriptional events downstream of functional TCR or TCR intermediates.
The four TCR gene clusters, α, β, γ, δ, are each composed of germline genes encoding discontinuous variable regions (Vα, Vβ, Vγ, Vδ), diversity regions (Dβ, Dδ), joining regions (Jα, Jβ, Jγ, Jδ), and constant regions (Cα, Cβ, Cγ, Cδ). The TCRγ locus is situated on human chromosome 7, and the remaining three are located on human chromosome 14, with the TCRδ locus embedded between the V α and J α regions of the TCRα locus. The TCRβ germline genes consist of approximately 50 Vβ, 2 Dβ, 13 Jβ, and 2 Cβ genes, whereas the TCRα germline genes consists of at least 70 Vα, 60 Jα, and a single Cα gene. The TCRβ and TCRα loci have many more genes than the TCRδ or TCRγ loci do; however, the diversity of TCR γδ T cells is increased by the splicing of 2 D δ regions to each other (VDDJ). Similar to the Ig genes in B cells, rearrangement of the TCR genes is carried out by the RAG1 and RAG2 proteins, with serial somatic rearrangements between the V, D, J, and C regions. Although N-region diversification is found in both the TCRα and TCRβ genes, T cells do not undergo somatic mutation as B cells do and instead undergo limited further rearrangements after emerging from the thymus.
Several major checkpoints regulate the development of TCR αβ and TCR γδ thymocytes. First, serial rearrangement of the TCR gene clusters in an ordered fashion ensures that the TCRβ and TCRα genes are not rearranged in developing TCR γδ T cells. Thus in human thymocytes TCRγ and TCRδ loci rearrange first, during the triple-negative stage. That human TCR γδ T cells generally have unrearranged TCRβ genes and that TCR αβ T cells generally have nonproductively rearranged TCRδ genes implies that only thymocytes that do not generate a functional γδ TCR go on to rearrange the TCRβ gene. The molecular mechanisms controlling this process are still being elucidated. The second or so-called beta selection checkpoint occurs after TCRβ rearrangement. Cells that undergo productive rearrangement of the TCRβ locus express functional TCRβ protein that complexes to the invariant pre-TCRα protein and CD3 signaling complex, thereby resulting in the expression of pre-TCR. In gene-deficient mice, where beta selection is known to occur during a precise phase of the triple-negative stage of development, loss of a number of signaling molecules or transcription factors downstream of pre-TCR results in defective beta selection, failure of differentiation into DP stage, and lack of proliferation or apoptosis (or both). Beta selection in humans begins during the CD34+ CD38+ CD1a+ stage and continues through the CD4 ISP and early DP stage of development (see Fig. 23-5 ). Thus only thymocytes that express functional TCRβ are allowed to proliferate and proceed to TCRα locus rearrangement.
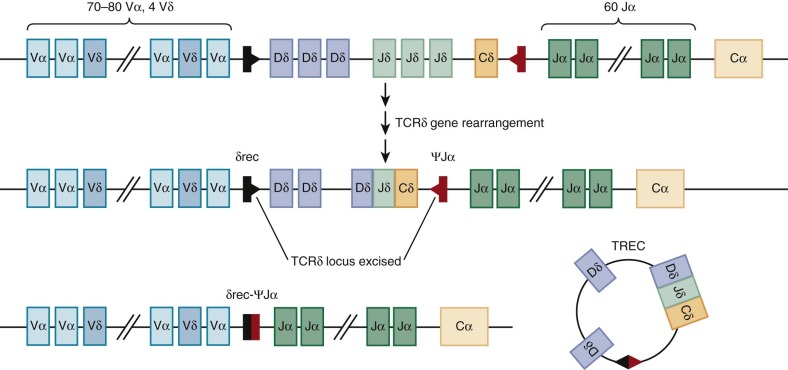
Before TCRα gene rearrangement, the TCRδ locus is excised by a nonproductive rearrangement between the δRec and ψJα regions that generates an episomal circle of DNA or T-cell receptor excision circle (TREC), which is increasingly being used on both a research and a clinical basis to quantify thymic activity ( Fig. 23-8 ). The utility of TREC as a marker of new T cell generation has recently been used to detect severe combined immunodeficiency (SCID) in newborn dried blood spots, and universal screening for SCID with TREC was endorsed for addition to the Recommended Universal Screening Panel in the United States by Secretary of Health and Human Services Kathleen Sebelius in May 2010. Unlike the TCRβ locus, rearrangement of the TCRα locus occurs processively; that is, Vα-Jα recombination of 5′ J segments that are nonproductive or out of frame is followed by further rearrangement on the same allele to more 3′ J segments. This process increases the chances of generating a complete TCR αβ protein from any single developing DP thymocyte. Successful rearrangement of the TCRα gene results in expression of the mature TCR αβ heterodimer and noncovalent association with CD3εγ, εδ, and ζζ dimer. This TCR complex appears during the late DP stage. These cells then undergo positive and negative selection (see the next section) and simultaneously commit to either the CD4 or the CD8 lineage to generate CD4 single-positive (SP) and CD8 SP thymocytes (see Fig. 23-5 ).
Positive Selection, Negative Selection, and CD4/CD8 Lineage Commitment
The massive proliferation of DP thymocytes results in TCR αβ thymocytes with an immense range of specificities, including many that are incapable of binding host MHC, cannot recognize host MHC complexed to endogenous peptide ligands, or have inappropriately high affinity for MHC–self-peptide and hence are autoreactive. Selecting only thymocytes with intermediate MHC–self-peptide affinity (so-called Goldilocks or just-right conditions) is critical for maintaining diversity of the repertoire while promoting self-tolerance.
Positive selection is the term used to describe the process of selecting thymocytes capable of recognizing host MHC complexed to self-peptides generated from the processing of endogenous proteins. DP thymocytes are exquisitely prone to cell death in response to γ-irradiation, corticosteroids, exposure to anti-CD3 antibodies, or simply removal of the thymus from the body, and thus a positive signal via TCR-MHC/peptide interactions is required to rescue the small number of DP thymocytes destined to become SP thymocytes from cell death. Gene-deficient mouse experiments have demonstrated that multiple elements of TCR signaling are required for optimal positive selection, including TCR components (TCRα, CD3δ), early signaling molecules (GADs, ZAP70), late signaling proteins (RasGRP, calcineurin, ERK), and transcription factors (nuclear factor of activated T cells [NF-AT], Egr family members). Interestingly, humans deficient in CD3δ and ZAP70 have been described who have discrepant development when compared with the knockout mouse counterparts. Deficiency of CD3δ in humans causes an earlier block in development at the CD4− CD8− stage, well before the development of DP thymocytes. ZAP70 deficiency results in selective deficiency of CD8 SP thymocytes and mature CD8+ T cells. Likewise, although in the mouse interaction of DP thymocytes with MHC on radioresistant cortical thymic epithelium is probably the dominant requirement for positive selection, the source of MHC and the relative contributions of MHC from thymic epithelium, thymic dendritic cells, or other thymic APCs to positive selection in humans are less clear.
Negative selection is the term used to describe the process of removing thymocytes that are self-reactive, also termed central tolerance, by clonal deletion. This is the first and major mechanism for control of autoimmunity, although clones that escape deletion in the thymus are subject to peripheral tolerizing influences (discussed later). In mice negative selection can be mediated by cortical epithelium, medullary epithelium, and medullary dendritic cells. However, it was never clear how thymocytes reactive against self-antigens expressed only in peripheral tissues, such as islet cells, were selected against in the thymus. In other words, how are these tissue-specific proteins expressed in the thymus? More recently, murine and human studies have revealed that all three cell types, medullary thymic epithelial cells (mTECs) in particular, exhibit promiscuous gene expression and transcribe a broad range of tissue-specific, nonthymic antigens. This insight helps explain how developing thymocytes reactive against nonthymic proteins can be centrally deleted during negative selection. A newly discovered gene, AIRE (autoimmune regulator), is a factor shown to control the transcription of tissue-specific antigen expression in murine mTECs. Simultaneously, this gene was shown to be responsible for the autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy (APECED) syndrome, characterized by chronic mucocutaneous candidiasis, hypoparathyroidism, adrenal insufficiency, and other organ-specific autoimmune manifestations. Thus this disease may be the first example of failure of negative selection underpinning a human monogenic autoimmune disorder.
Maturation of CD4 and CD8 SP thymocytes from DP thymocytes occurs concurrently with positive selection and results in “matching” of coreceptor to MHC restriction with downregulation of the opposite coreceptor. The mechanisms controlling the CD4/CD8 lineage decision are separable from those governing positive selection and have been the subject of active investigation in murine models. Patients with defects in a number of genes that control MHC class II expression, including RFXANK, RFX5, RFXAP, and CIITA, all manifest the so-called bare lymphocyte syndrome , a rare autosomal recessive disorder characterized by decreased numbers of CD4+ T cells, deficient helper function, and absence of specific antibody production because of lack of CD4-mediated B-cell maturation.
The Spleen
Historical Perspective
A complete understanding of the adaptive immune system requires a discussion of the structure and function of the spleen, a critical secondary lymphoid organ with both immunologic and nonimmunologic functions. Since ancient times the spleen has occupied a mysterious niche among organs within the human body. Its proposed functions and necessity for good health were subjects of debate and discussion for centuries. Hippocrates believed that the spleen helped balance the body’s “humors,” a process necessary to promote harmony and stability. Plato and Aristotle further suggested, on the basis of its location, that it assisted or balanced the liver. Galen noted the anatomic connections among the spleen, liver, and stomach and concluded that the spleen had true digestive functions that included the filtering of humoral impurities. Subsequently, the spleen gained a reputation for controlling feelings and emotions, including laughter, ill temper, melancholy, and others. Shakespeare often referred to the spleen as an organ of anger, merriment, or other strong emotion. During the Renaissance, when the study of anatomy and physiology became popular, the spleen’s microscopic patterns supported a role for filtration; however, only circulatory connections between the spleen and the stomach, liver, and other digestive organs could be identified. Moreover, survival of both animals and humans after surgical splenectomy demonstrated that the spleen was not absolutely required for life. Only in the modern era, with the discovery of individual blood cells, have investigators begun to elucidate the true functions of the spleen. We now recognize that the spleen has vital roles in blood filtration and immunologic competence.
Anatomy and Physiology
The spleen can be identified in human embryogenesis during week 5 of gestation, although the development of characteristic splenic architecture with influx of red and white blood cells occurs much later in gestation. At birth the spleen weighs about 10 g, then 30 g at 1 year of age, 60 g by 5 years, and approximately 200 g in adults. Located in the left anterior aspect of the rib cage, the spleen can have a triangular, crescent, or rhomboidal shape. Spleen size is usually estimated clinically by how far below the left costal margin the tip of the spleen can be palpated. Frequently, an enlarged spleen is most easily palpated at the midclavicular line, but it can extend prominently toward the midline or even to the midaxillary line in some patients.
The spleen receives 3% to 5% of the total cardiac output, thus making it a highly perfused organ. Blood enters and leaves the spleen primarily through the splenic artery and vein, respectively, both located at the hilum on the concave (medial) side of the organ. Within the splenic parenchyma, the splenic arterial branches have little overlap, thereby allowing classification of the spleen into lobes and segments separated by relatively avascular planes.
Histologically, the spleen can be separated into two distinct regions, the red pulp and the white pulp, so named because of the blood cells most commonly found in each area. These two regions provide the vital functions of blood filtration and immunologic response, respectively, which are made possible through a unique circulation pattern found within the spleen. Figure 23-9 illustrates the arterial and venous circulations of the spleen and the close proximity of the red and white pulp.
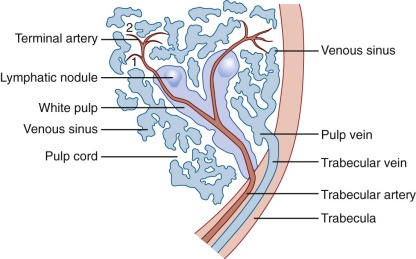
The white pulp consists primarily of collections of lymphocytes and macrophages that form small white nodules (1 to 2 mm in diameter) distributed throughout the splenic parenchyma. As the largest arteries within the spleen branch progressively into the smallest arterioles, the white pulp can be recognized as a circumferential ring of leukocytes that surround the vessel, also known as the periarterial (or periarteriolar) lymphoid sheath (PALS). Whereas blood cells tend to continue within the vessel, plasma elements enter the PALS by way of tiny vessels that arise perpendicular to the flow of blood. Soluble antigens are thus herded into the white pulp, where they encounter dendritic cells and macrophages that function as professional APCs and allow proper immune stimulation of T and B lymphocytes.
The PALS consists primarily of T lymphocytes, and this cylindrical cuff of cells becomes thinner as the artery becomes smaller. Spheroidal lymphoid follicles arise at intervals from the PALS, usually where arterial branch points occur, and these follicles contain primarily B lymphocytes ( Fig. 23-10 ). The primary follicles contain naïve mature B cells that co-express IgM and IgD; secondary follicles contain activated B cells within its germinal center (GC), all termed follicular B cells. Naïve follicular B cells in the primary follicle are CD27−, in contrast to memory B cells, discussed later, which express CD27. In the mouse T cells congregate around the central arteriole in the so-called PALS studded by follicles and GCs containing B cells; the marginal zone delineates the white pulp from the red and surrounds both the follicles and PALS. In humans, by contrast, T cells and B cells tend to interdigitate, and the follicles are encompassed within the PALS. The marginal zone in humans is positioned around the follicles, does not envelop the PALS, and is not in direct contact with red pulp. Marginal-zone B cells are IgM+ IgD− CD1c+ CD23− and thus are distinguished from follicular B cells (IgM− IgD+ CD1c− CD23+). Additionally, marginal-zone B cells express high levels of CD21 and as a result are highly responsive to complement. Finally, in contrast to rodent marginal-zone B cells, which are sessile, marginal-zone-like B cells in humans can circulate in blood and have a memory phenotype, being IgM+ IgD+ CD27+.
The red pulp represents a large open circulation within the spleen through which erythrocytes pass slowly for filtration and culling functions. The red pulp contains the sinusoids, which are lined by macrophages, and the splenic cords, which lie between the sinusoids and are composed of connective tissue, epithelial cells, monocytes, and macrophages. After plasma is redirected toward the white pulp, the blood cells that continue through the splenic circulation are mostly erythrocytes, and they form the red pulp as they exit the arteries. Occasionally, erythrocytes pass directly from the arterial to the venous circulation by way of traditional endothelialized capillaries, a process known as the closed (rapid) splenic circulation , which is primarily nutritive. Much more commonly, however, erythrocytes leave the arterial circulation and empty into large pools called the splenic cords , where they encounter macrophages and other immune effector cells. Erythrocytes must survive these intimate encounters in this hypoxic and acidic environment and then pass from the cords through an endothelialized barrier into the splenic sinuses and finally back to the venous circulation. This open (slow) circulation allows careful filtration of individual erythrocytes, with repair or phagocytosis of cells that cannot successfully negotiate the return trip.
Important Functions
The spleen provides several important immunologic functions. Skimming of plasma into the PALS allows particulate antigens to interact with dendritic cells and other APCs, which initiates the immune response for T- and B-lymphocyte response and proliferation. Polysaccharide antigens in particular require this unique immunologic environment for optimal antibody response. Intravenous antigens, such as those found during infection or after immunization, are primarily processed by the spleen. Not surprisingly, normal splenic function is required for optimal early (IgM) response to most intravenous antigens, as well as the secondary (IgG) response. In contrast, antigens administered by the intramuscular or subcutaneous routes do not require normal splenic function for a robust immune response. Finally, bloodborne bacteria, particularly encapsulated organisms, are removed by splenic macrophages located in the PALS and in the cords. Without a functioning spleen, patients are susceptible to overwhelming bacterial sepsis and exaggerated parasitemia from Plasmodium, Babesia, or other organisms. The interaction between specialized T helper cells and follicular B cells in the GC is further described in later sections.
The spleen also has nonimmunologic functions. All RBCs pass through the spleen many times each day. The spleen serves as an efficient filter for erythrocytes and is able to perform grooming (removal of antibodies or other surface molecules), culling (destruction of cells), and pitting functions (removal of intracellular material). While moving slowly through the hostile environment of the splenic cords, erythrocytes are carefully examined for abnormalities, a process constituting a biologic form of quality control. Macrophages in the red pulp identify abnormal erythrocytes, such as those that have IgG or complement on their surface, and either remove a portion of their membrane or fully engulf them. Newly made reticulocytes spend their first day or two in the spleen, during which the cells appear to be groomed with removal of cytoplasmic organelles and surface adhesion molecules. Normal mature erythrocytes, in contrast, typically endure macrophage scrutiny without difficulty. To leave the red pulp, erythrocytes must then pass back from the splenic cords through potential spaces in the endothelial lining of the splenic sinuses. To negotiate these tiny apertures, known as the cords of Billroth , the erythrocytes must deform themselves substantially ( Fig. 23-11 ). Cordal macrophages phagocytose erythrocytes that cannot deform themselves sufficiently, such as senescent cells; as erythrocytes age, they lose volume, gain density, and eventually lose the deformability that allows reentry into the splenic sinus. In certain pathologic conditions, such as immune-mediated hemolytic anemia, as well as a variety of erythrocyte membrane, hemoglobin, and enzyme disorders, younger erythrocytes will also be trapped in the splenic cords. Erythrocytes with intracellular inclusions, such as nuclear remnants known as micronuclei (Howell-Jolly bodies), denatured hemoglobin (Heinz bodies), siderotic granules (Pappenheimer bodies), and malarial parasites, have their inclusions “pitted” out before they pass through the endothelial slits and return to the circulation.
To a much lesser extent, the human spleen can also serve as a reservoir for circulating blood cells. Approximately 50 mL of blood is usually contained in a normal spleen, as well as 30% of the platelet mass. In the abnormal state (discussed later), the spleen can enlarge and trap a larger portion of the circulating blood cells, a condition known as hypersplenism . The spleen may also serve a small role in iron metabolism and homeostasis. Unlike the spleen in other species, however, it has no appreciable role in human hematopoiesis.
Abnormalities of the Spleen
Congenital Asplenia and Polysplenia
Congenital asplenia is found in children with the rare Ivemark syndrome, in which the body has bilateral “right-sidedness” with trilobed lungs and a centralized liver. Cardiac defects are common, as well as a risk of infection. In even rarer cases patients have congenital asplenia with no other abnormalities. This resembles mice with a targeted deletion of Hox11 (also called Tlx1 ). No defects in Hox11 have been discovered in humans with isolated congenital asplenia. Instead, about half of the patients have a heterozygous inactivating mutations in RPSA (ribsomal protein SA), a component of the small subunit of the ribosome. How haploinsufficiency of a part of the ribosome, which is needed in all cells, specifically blocks development of the spleen remains a mystery. A heterozygous missense mutation in Nkx2-5 has also been found in one kindred. This makes sense because Nkx2-5 interacts with Pbx1 and p15Ink4b to regulate spleen organogenesis in mice.
A number of genes in humans and mice are linked to asplenia combined with multiple defects in other tissues. Examples include: Nkx2-3, Nkx3-2 (or Bapx1), Sox11, Wt1, Pbx1, Tcf21 (or Pod1), and HuR. Interestingly, HuR interacts with a putative regulatory element in the Hox11 gene.
Uptake of radionuclide by functioning splenic tissue can be difficult to document because the anatomic features are distorted and liver tissue can obscure any splenic uptake. Circulating erythrocytes with Howell-Jolly bodies or intracellular vesicles that appear as pits or pocks allow the diagnosis of asplenia to be established noninvasively. Figure 23-12 illustrates the Howell-Jolly bodies that are found in the peripheral blood smear of an asplenic person. The converse arrangement of bilateral “left-sidedness” leads to congenital polysplenia with several spleens of varying size. In most patients the splenic tissue in polysplenia has normal function. Cardiac defects are common, as well as hepatobiliary abnormalities.
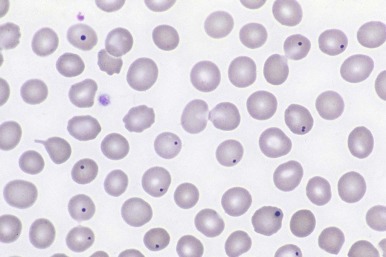
Accessory spleens can be identified in 15% of normal persons. In most of these individuals, a single accessory spleen measuring only 1 to 2 cm in diameter is present either in the splenic hilum or in the tail of the pancreas. Multiple accessory spleens can exist, however, as well as ectopic spleens in locations ranging from the abdomen to the pelvis and scrotum. The function of accessory spleens depends on the quality and quantity of blood flow; in most instances their filtration capabilities are minimal, but antibody responses may be present.
Splenosis and Splenoptosis
Splenoptosis, or wandering spleen, occurs when the spleen is not fixed within the retroperitoneum. The resulting mass can be palpated anywhere in the abdomen and may require reattachment (splenopexy) or even splenectomy if symptoms of sequestration or torsion develop. Splenosis refers to the autotransplantation of splenic tissue into the omentum or peritoneal surfaces of the abdominal cavity. Splenosis typically occurs after fracture or rupture of the spleen as a result of spillage and subsequent implantation of splenocytes. Although some protection against infection may be afforded by splenosis, filtering functions and immunologic responses are limited by the relatively small total amount of splenic tissue and reduced blood supply.
Splenic Sequestration
Sequestration refers to enlargement of the spleen that occurs when blood enters the organ but is unable to exit properly. This condition is most often observed in children with sickle cell anemia when venous return of blood is hindered by intrasplenic sickling of erythrocytes within the red pulp. Splenic sequestration also occurs in other settings with abnormal erythrocytes, however, including congenital spherocytosis. Splenic sequestration can develop acutely with sudden pooling of blood within the spleen and result in rapid and painful expansion of the splenic parenchyma and capsule. Children exhibit pallor, fatigue, severe anemia, and occasionally hypovolemic shock. Occasionally, splenic sequestration develops over a period of days or weeks and is then termed subacute or chronic because it is accompanied by relatively asymptomatic splenomegaly.
Hypersplenism
Hypersplenism refers to the condition of splenomegaly with non–immune-mediated trapping of peripheral blood cells and results in mild to moderate cytopenia. Hypersplenism can develop in patients with abnormal erythrocytes, metabolic storage disorders, or other causes of chronic congestive splenomegaly. In these clinical settings, the spleen first enlarges because of progressive trapping of erythrocytes or storage material and then subsequently becomes a reservoir for all circulating blood cells. Typically, the peripheral platelet count is lower than normal, but the leukocyte count can also be low. If surgery is required, peripheral blood counts normalize after splenectomy.
Surgery Involving the Spleen
Although the spleen has important hematologic and immunologic functions, in some instances surgical removal is beneficial. A variety of congenital anomalies of the spleen, as well as numerous pathophysiologic processes, are indications for splenectomy.
Benign tumors of the spleen are relatively rare in childhood but primarily include congenital cysts (epidermoid or lymphangiomatous) or pseudocysts that result from liquefaction of splenic parenchyma after infarction or hemorrhage. These tumors may require splenectomy, depending on their size. Malignant splenic tumors are almost always lymphomatous or leukemic in origin, with cancerous cells found diffusely or in a nodular pattern throughout the splenic parenchyma. Perhaps somewhat surprisingly, very few solid tumors metastasize to the spleen. As a general guideline, splenectomy is not usually indicated in the setting of malignancy.
Splenomegaly requiring splenectomy can develop in a variety of clinical settings, including metabolic storage disorders such as Gaucher disease, transfusion-acquired iron overload from chronic erythrocyte transfusions, and splenic congestion resulting from portal hypertension. Commonly, splenectomy is the recommended therapeutic option for splenic enlargement in patients with primary erythrocyte abnormalities. Congenital hemoglobin, enzyme, and membrane disorders lead to trapping of the abnormal erythrocytes within the splenic cords and gradual expansion of the red pulp. In these settings splenectomy can relieve the signs and symptoms of splenomegaly, including abdominal discomfort, anemia, hypersplenism, and dependence on transfusions.
Total surgical splenectomy is generally performed by open laparotomy by way of a left subcostal incision. Laparoscopic splenectomy has become a popular and safe alternative in children but requires careful attention and may be technically difficult with very large spleens. With the laparoscopic approach, small amounts of residual splenic tissue may be left or accessory spleens may be missed, which can be a problem for patients with immune-mediated hematologic disorders. When splenectomy is recommended for autoimmune hemolytic anemia (AIHA) or immune thrombocytopenic purpura (ITP), all splenic tissue must be removed to prevent recurrent autoantibody production. Over the past decade subtotal (partial) splenectomy has become a viable therapeutic alternative to total splenectomy. The partial splenectomy approach is possible because the spleen can be divided into distinct lobes and segments. Removal of 80% to 90% of the enlarged spleen usually provides relief from splenomegaly while retaining sufficient splenic tissue for immune competence. Partial splenectomy has been used successfully in children with nonparasitic splenic cysts, hereditary spherocytosis, homozygous β-thalassemia and sickle cell disease, although splenic regrowth may limit its long-term efficacy. Even children with massive splenomegaly can successfully undergo partial splenectomy. Animal studies suggest that a third of the normal spleen mass is required for normal plasma filtration and antibody formation. Compared with splenic tissue reimplanted either by surgical autotransplantation or splenosis, partial splenectomy provides superior retained immune function. Laparoscopic partial splenectomy combines these two new approaches and may represent an ideal option for selected patients who require surgical splenectomy; it is becoming more commonly available for children.
The primary danger after total splenectomy is fatal bacterial sepsis, sometimes referred to as overwhelming postsplenectomy infection (OPSI). First recognized more than 50 years ago, bacteremia evolving rapidly into fatal sepsis with or without meningitis can abruptly develop in a patient without splenic function. Pathogens associated with OPSI are primarily encapsulated bacterial organisms and include Streptococcus pneumoniae, Haemophilus influenzae type b, and Neisseria meningitidis. The presumed pathophysiologic origin of OPSI is loss of plasma filtration by the spleen, which allows bacteria to multiply within the bloodstream and evolve from low-grade bacteremia to fulminant sepsis. Risk factors for OPSI include younger age at initial clinical evaluation, younger age at splenectomy, and failure to comply with prophylactic antibiotics or vaccinations. The true risk for the development of OPSI has been estimated in two large studies, although many of the patients were from the prevaccination era. First, in a literature review covering 35 years from 1952 to 1987 (5902 patients), the incidence of infection after splenectomy in children younger than 16 years was 4.4% with a mortality rate of 2.2%; for adults the corresponding values were 0.9% and 0.8%, respectively. Children younger than 5 years and especially infants had a much higher risk for sepsis. In a later survey of 226 splenectomized patients with hereditary spherocytosis that included 5461 years of postoperative follow-up, the estimated mortality rate was 0.73/1000 years for the entire group, but somewhat higher for children. The introduction of polysaccharide and now protein-conjugated vaccines has greatly helped decrease the incidence of pneumococcal bacteremia and sepsis and will help offset the lack of splenic filtration function for children undergoing splenectomy. However, even with the use of presplenectomy immunization, prophylactic antibiotics, and prompt medical attention for fever, a splenectomized child still has a finite risk of OPSI developing with substantial morbidity and mortality.
B-Cell Activation and Function
An appropriately diverse population of naïve B cells should collectively be capable of responding to any soluble antigen that the individual encounters, yet there are very few naïve B cells that are capable of recognizing any given antigen. B-cell activation that occurs in the appropriate costimulatory context is the trigger for clonal expansion of the responding B cell. A portion of these activated B cells differentiate into memory cells, which rapidly respond on reencounter with the same antigen, and another portion into plasma cells, which continuously secrete antibodies (see Fig. 23-2 ). In addition, activated B cells undergo isotype switching from low-affinity IgM to high-affinity IgG, IgA, or IgE and somatic hypermutation, also called affinity maturation , thus further ensuring that the humoral response is exquisitely specific, effective, and long lasting. Recognition of antigen by the BCR occurs with or without T-cell help, depending on the type of antigen, so-called thymus-dependent (TD) or thymus-independent (TI) antigens. BCR signals that occur during TD responses are modulated and enhanced by cell-cell contact with T cells by way of critical costimulatory molecules, discussed further in the section on T-cell costimulation.
Thymus-Dependent Versus Thymus-Independent B-Cell Responses
Unlike T cells, which recognize either short peptides or small lipid molecules that must be presented in the cleft of an MHC or MHC-like molecule, the BCR is capable of binding a variety of antigens, including soluble proteins and polysaccharides. Recognition of protein antigen generally requires cooperation with T cells, or T-cell help, and thus such antigens are TD antigens. Naïve follicular B cells in lymph node follicles or the spleen recognize their cognate antigen while contacting a helper T cell specific for the same antigen; linked T-B interaction then fuels the formation of a key structure, the GC, where isotype switching, somatic hypermutation, memory cell generation, and plasma cell differentiation occur.
TI antigens are classified as type 1 or type 2 (TI-1 and TI-2). TI-1 antigens are bacterial components that can trigger B-cell activation regardless of Ig specificity and thus lead to polyclonal activation. These components fall into the general category of pathogen-associated molecular pattern (PAMP) molecules, the canonic example being lipopolysaccharide (LPS). PAMP molecules activate B cells by binding to a distinct family of receptors, the Toll-like receptors (TLRs), thereby accounting for their independence from Ig. The Ig secreted in response to TI-1 antigens is typically low-affinity IgM because TI-1 antigen binding does not induce class switching.
TI-2 antigens, in contrast, are molecules that have a repetitive structure and activate B cells by cross-linking of multiple BCRs on a single cell, hence leading to specific clonal antibody production. These antigens are typically bacterially derived polysaccharides, such as those found in the coating of encapsulated organisms. Marginal-zone B cells are important for responses to TI-2 antigens, and the fact that marginal-zone B cells typically take years to mature in children probably underlies the lack of response of children younger than 2 years to polysaccharides. The spleen is the major site of TI-2 antigen recognition, and the antibody produced again is often IgM, although IgG responses also clearly occur. Class switching in this instance does not depend on cognate T-cell help but probably is enhanced by noncontact T-cell factors such as the cytokine interleukin-4 (IL-4).
B-Cell Receptor Triggering and Signaling
B-cell activation involves a complex array of molecular signaling events. The signaling cascades that emanate from the BCR are highly analogous to those triggered in T cells by the TCR and involve a host of kinases, adaptor proteins, and other enzymes.
In brief, B-cell activation commences with engagement of surface Ig, which initiates receptor oligomerization, an event that depends on B-cell coreceptors such as the stimulatory CD19 and CD21 receptors and the inhibitory low-affinity FcR. In addition, aggregation of Ig receptors takes place in specialized membrane microdomains, termed lipid rafts, that are characterized by their cholesterol- and sphingolipid-rich content. In cellular systems lipid rafts have been postulated to function in intracellular trafficking, apical sorting, and signal transduction. In both T and B lymphocytes, lipid raft components may function as a potential signaling platform to foster the aggregation and co-localization of signaling proteins.
Within the lipid raft, the Ig molecule transmits signals through the associated Ig-α and Ig-β molecules ( Fig. 23-13 ), together termed the BCR; the BCR, like other members of the immune recognition receptors, has no intrinsic tyrosine kinase activity. On oligomerization the cytoplasmic domains of Ig-α and Ig-β become tyrosine phosphorylated on so-called immunoreceptor tyrosine–based activation motifs (ITAMs) with the amino acid consensus sequence D/ExxYxxL/Ix7YxxL/I. Phosphorylation of ITAMs is mediated by Src kinase family members, notably Fyn, Blk, and Lyn (see Fig. 23-13 ), which then recruit the critical tyrosine kinase Syk by way of its Src homology 2 (SH2) domains. Syk itself, when bound, requires activation either by autophosphorylation or phosphorylation by Src kinases, and thus clustering of multiple ITAM-bearing BCRs, Src kinases, and Syk is required to achieve a threshold of activation that initiates downstream cascades.
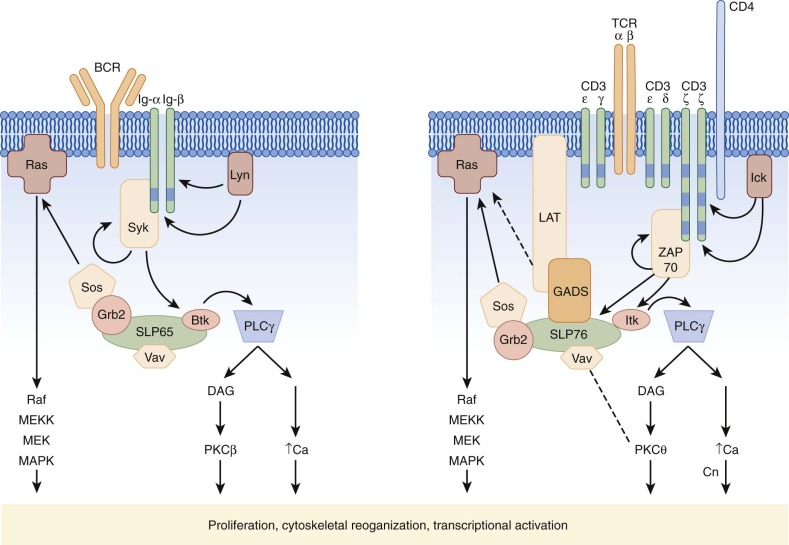
Activation of Syk, similar to activation of ZAP70 in T cells (see later), is a crucial linchpin in BCR signaling. Syk itself functions as a tyrosine kinase and is capable of phosphorylating key substrates, which then activate three main signals, the protein kinase C (PKC), intracellular calcium, and Ras pathways. Activation of these pathways occurs by assembly of a macromolecular complex of critical signaling molecules clustered on an adaptor protein known as the B-cell linker protein (BLNK) or SLP65. Recruitment of a member of the Tec kinase family, Btk, to SLP65 by way of the SH2 and Src homology 3 (SH3) domains and to the membrane by the Pleckstrin homology (PH) domain activates Btk. The critical nature of Btk activation in BCR signaling is underscored again by its role as the gene responsible for X-linked agammaglobulinemia. Btk in turn activates an isoform of phospholipase C (PLCγ2) that hydrolyzes inositol phosphates to generate diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP 3 ). DAG activates both an isoform of PKC (PKCβ) and Ras, whereas IP 3 induces an increase in intracellular calcium. Recruitment of Sos by way of the adaptor protein Grb2 also activates the Ras pathway, as does Vav, a guanine nucleotide exchange factor for Rac. Together, activation of these three pathways then triggers activation of mitogen-activated protein kinases (MAPKs); reorganization of the actin cytoskeleton; and nuclear translocation of critical transcriptional activators, including the NF-κB family, that promote survival and clonal expansion (see Fig. 23-13 ).
Coreceptors Involved in B-Cell Activation
Three important coreceptors for B-cell activation are CD21 (also called complement receptor 2 [CR2]), CD19, and CD81. Antigens derived from bacteria or other extracellular microorganisms are more efficiently cleared when opsonized or bound by complement components, a mechanism that links the early innate immune response elicited by activation of complement to the adaptive humoral B-cell response. Binding of C3b to CD21 coclusters CD21, CD19, and CD81 with the BCR within lipid rafts. Phosphorylation of CD19 by proximity to the activated BCR complex in turn generates docking sites for phosphatidylinositol 3′-kinase (PI3K), for Vav, and for Lyn. PI3K generates phospholipid intermediates that recruit Btk, as described earlier. Vav activates Rac, thereby activating MAPKs and inducing cytoskeletal reorganization. The response to opsonized antigens recognized by the BCR via its specific epitope is thus greatly enhanced by the binding of C3b to CD21. CD81 appears to function primarily as an adaptor and is critically required for BCR and CD21/CD19 to associate with lipid rafts.
Stimulation by way of the antigen receptor alone is insufficient to activate B or T cells, and, in fact, binding of receptor-ligand pairs between B and T cells is critical for activation of both. Many costimulatory pathways are shared between B and T cells and are discussed further later; binding of CD40 to its ligand CD40L (CD154) is the most crucial of these pathways for B-cell activation. CD40 is a 48-kd member of the TNF receptor superfamily that is expressed on B cells and other APCs constitutively, whereas CD40L is a 33-kd transmembrane protein with homology to TNF upregulated on activated CD4+ T cells. CD40/CD40L binding has been shown to be essential for B-cell growth and differentiation. In vitro stimulation of B cells by cross-linking by way of monoclonal antibodies is sufficient to promote growth and class switching and is synergized by BCR stimulation or treatment with IL-4, or both. Engagement of CD40 with trimeric CD40L and clustering of the cytoplasmic tails lead to recruitment of a class of adaptor proteins, the TNF receptor–associated factors (TRAFs), which in turn activate a variety of kinases that ultimately culminates in activation of MAPK and NF-κB. In addition to clonal expansion and survival, CD40 signaling also leads to upregulation of other costimulatory molecules such as CD80 (B7) and CD54 (intercellular adhesion molecule 1 [ICAM-1]).
The relatively recently discovered TLR can trigger B-cell activation alone (see earlier discussion) and also can serve a costimulatory function with BCR signals. At least 11 TLRs in humans have been found, each recognizing different microbial components, such as LPS (via TLR4), peptidoglycan (TLR2), double-stranded RNA (TLR3), flagellin (TLR5), and CpG DNA (TLR9). The response to intact microbes may involve recognition of microbial components simultaneously by the BCR and TLR, and interestingly, BCR-TLR co-recognition may play a role in the generation of autoantibodies.
Follicular B-Cell Differentiation: the Germinal Center
Activation of B cells and differentiation into memory and plasma cells have become increasingly well understood in the context of cellular trafficking and the complex architecture of the specialized anatomic site, the GC. B cells, a critical subset of CD4+ T cells (follicular helper T cells or T FH ), and specialized follicular dendritic cells (FDCs) must interact through costimulatory contact and soluble factors to generate a long-lasting humoral response.
In the early stages of infection, innate immune cells such as dendritic cells and macrophages capture antigen, migrate to lymph nodes, and present peptides in the cleft of class II MHC to CD4+ T cells. Simultaneously, naïve B cells that recognize the same pathogen can internalize complex protein particles by receptor-mediated internalization of Ig, with the protein fragments being broken down and displayed as peptides by class II MHC. Naïve B cells that encounter their cognate antigen and become activated then migrate through the T-cell zone and contact T cells stochastically during the journey. When T and B cells that recognize the same antigen interact through TCR-MHC interactions, costimulatory signals, namely CD40-CD40L interactions, enhance the interaction and result in clonal expansion of both T and B cells. These activated “helped” B cells then travel to the follicle and infiltrate a network of FDCs. FDCs are thought to have a specialized function consisting of capture of immune complexes and simultaneous binding of FcR to antibody and complement receptor 3 (CR3) to components of complement. The antigen component of the immune complex can then be displayed to passing B cells by way of the BCR. Activated B cells that recognize antigen displayed on FDCs proliferate rapidly after further costimulation by CD4+ T cells and become the centroblasts and centrocytes that form the GC.
The role of T FH in this process has been increasingly elucidated in recent years. These specialized CD4+ T cells were first identified on the basis of their expression of CXCR5, a receptor typically found on B cells that binds the B cell homing chemokine CXCL13. They can be found in spleen, tonsil, and lymph node, optimally positioned in the B follicles where they function, and phenotypically similar cells circulate in the blood. Functionally T FH are the key contributors to B cell help in the secondary lymphoid organs by virtue of the high-level production of IL-21, a pleiotropic cytokine that is critical for induction of class switching along with IL-4, and high surface expression of ICOS. The development of T FH depends on IL-21 itself and the master transcription factor BCL6, as further discussed in the section on helper T cell differentiation and function.
Somatic hypermutation is thought to occur at the centroblast stage and further diversifies the overall antibody pool by introducing mutations in the V H and V L regions of Ig in B cells involved in a GC response. Nucleotides are thought to be mutated in a somewhat random fashion, in both the rearranged and nonrearranged V regions, but ultimately survival of B cells that have undergone hypermutation is dictated by positive and negative selection based on the affinity of the expressed Ig. In other words, mutations that disrupt affinity to the antigen to which the animal is responding will be selected against, whereas those that enhance affinity will be selected for, hence the term affinity maturation .
Isotype or class switching is thought to occur at the centrocyte stage. Because all naïve B cells express IgM, a low-affinity antibody, inducing activated B cells to produce high-affinity IgG or IgA significantly enhances the functional humoral immune response to specific antigen at the time of infection and requires splicing of the V(D)J regions to the C γ or C α region. CD40-CD40L interactions are critical for this process, and T-cell–derived cytokines enhance class switching while influencing the Ig class or subclass. In the mouse, T-B interactions in the context of interferon-γ (IFN-γ) tend to promote switching to IgG2a and IgG3, whereas IL-4 directs switching to IgG1 and IgE (see Table 23-2 ).
| Class | Isotype | Heavy Chain | Molecular Formula | Molecular Mass (kd) | Plasma Concentration (mg/mL) * | Cross Placenta | Fix Complement | Biologic Functions |
|---|---|---|---|---|---|---|---|---|
| IgG | Complement activation, opsonization for Fc receptor | |||||||
| IgG1 | γ1 | γ1 2 κ 2 γ1 2 λ 2 | 150 | 7-9 | + | + | Antitoxins, antibody to most bacterial antigens | |
| IgG2 | γ2 | γ2 2 κ 2 γ2 2 λ 2 | 150 | 2-3 | + | + | Antipolysaccharide antibodies to bacterial capsules | |
| IgG3 | γ3 | γ3 2 κ 2 γ3 2 λ 2 | 150 | 0.7-1 | + | + | Cytophilic antibodies (e.g., to Rh antigens) | |
| IgG4 | γ4 | γ4 2 κ 2 γ4 2 λ 2 | 150 | 0.3-0.5 | + | − | Anticoagulants, such as anti–factor VIII in hemophiliacs | |
| IgM | µ | (µ 2 κ 2 ) 5 (µ 2 λ 2 ) 5 | 950 | 1.5 | − | + | Receptor in B-lymphocyte membrane, antibodies to lipopolysaccharides, complement activation, natural antibody | |
| IgA | 150 or 300 | Present in external secretions, alternative complement activation (?) | ||||||
| IgA1 | α1 | α1 2 κ 2 α1 2 λ 2 | 1-3 | − | − | Virus-neutralizing antibody | ||
| IgA2 | α2 | (α2 2 κ 2 ) 2 | 0.5-0.7 | − | − | Principal secretory antibody | ||
| IgD | δ | δ 2 λ 2 δ 2 κ 2 | 180 | 0.3 | − | − | Unknown, receptor in B lymphocyte membrane | |
| IgE | ε | ε 2 λ 2 ε 2 κ 2 | 190 | 0.0005 † | − | − | Anaphylactic antibodies or “reagins,” binds to mast cells, mediator of release |
* Plasma concentrations of the immunoglobulins vary significantly with age in pediatric populations. Adult norms are given here. IgG at birth reflects maternally transferred immunoglobulin and wanes over several months, with a nadir at approximately 4 months and rising thereafter as the infant’s own production takes over. IgM and IgA levels rise with age during infancy and childhood.
† Usually expressed as international units; up to 200 IU is considered normal.
GC B cells then differentiate into either plasma cells or switched memory B cells. How GC B cells “choose” between the plasma cell and memory B-cell fate remains unclear. Plasma cells are terminally differentiated B cells found primarily in bone marrow and are specialized for high-level secretion of IgG, IgM, and IgA. They have a characteristic histologic appearance with so-called clockwork chromatin, eccentric nuclei, and markedly expanded endoplasmic reticulum and can be identified by expression of CD138 (also called syndecan-1). Class-switched memory B cells, which generally can be identified by virtue of the loss of IgD and gain of CD27 surface expression, are able to circulate back to bone marrow and secondary lymphoid organs and, like memory T cells discussed later, are responsible for the rapid and effective anamnestic immune response characteristic of acquired immunity. A number of congenital deficiencies that affect T-cell–dependent B-cell maturation are characterized by a lack of GC formation and switched memory B cells; such conditions include CD40L deficiency (discussed later), X-linked lymphoproliferative disease (caused by mutation in SH2D1A or SLAM-associated protein [SAP]), and two molecules recently shown when mutated in humans to cause common variable immunodeficiency, ICOS (inducible T-cell costimulator) and TACI (transmembrane activator and calcium modulator and cyclophilin ligand interactor). More recently, both somatic hypermutation and class switch recombination have been found to require a newly discovered activation-induced deaminase (AID) that deaminates deoxycytidine to form deoxyuridine; subsequent repair of the deoxyuracil-deoxyguanosine mismatch triggers nucleotide substitution or excision, thereby generating diversity and also joining VDJ regions to a different constant region class. Humans born with defects in the AID gene have failure of both processes that results in a hyper-IgM phenotype.
B-Cell Tolerance
Like T-cell tolerance (discussed later), B-cell tolerance is essential to prevent autoimmune reactions. Immature B cells can be made tolerant in the bone marrow to antigens expressed by the host (i.e., self-antigens) through clonal deletion. If the surface IgM (sIgM) expressed on an immature B cell binds with high avidity to a self-antigen, the cell will be stimulated to undergo apoptosis, a form of cell death characterized by chromosomal fragmentation and membrane blebbing. The B cell may also undergo receptor editing, a process whereby that B cell’s Ig gene will further rearrange to express a different, nonautoreactive Ig molecule. The few B cells that escape central deletion may be rendered anergic in the periphery, a state in which the B cell expresses a gene that encodes a self-reactive antibody yet is unable to respond to antigen. The molecular mechanisms responsible for anergy are incompletely understood but may involve alternative signaling when the BCR is activated in the presence or absence of costimulation. For instance, BCR triggering in the context of concurrent TLR signaling (secondary to microbial products in addition to the BCR-specific antigen) leads to upregulation of CD86, which engages CD28 on T cells and recruits T-cell help. In the absence of TLR signals, the upregulation of CD86 is not sustained, and the BCR signals are attenuated. A group of TNF and TNF receptor family members, including BAFF (also known as BLyS, TNFSF13B ) and APRIL ( TNFSF13A ), whose receptors include TACI ( TNFRSF13B , CD267), BCMA ( TNFRSF17 , CD269) and BAFF-R ( TNFRSF13C , CD268), have been shown to be important in B-cell homeostasis and autoimmunity. Interestingly, mice that overexpress BAFF exhibit autoimmunity, and humans with a variety of rheumatologic diseases have been shown to have high BAFF serum levels. The increasing use of rituximab, humanized anti-CD20 antibody, for the treatment of autoimmune disease and, recently, chronic graft-versus-host disease (GVHD) underscores the importance of dysregulated humoral immunity in human disease. More recently, a fully human IgG1-λ monoclonal antibody specific for BAFF/BLyS, belimumab, has been shown to have efficacy in systemic lupus erythematosus and is approved by the U.S. Food and Drug Administration (FDA) for treatment of mild to moderate disease.
Function of Secreted Immunoglobulin
Secretion of a diverse repertoire of Ig is essential for protection of the host from infection and for neutralization of toxin. Once formed, binding of specific antibodies to microorganisms, virally infected cells, toxins, or other pathogens is essential for complement-mediated lysis, antibody-dependent cellular cytotoxicity (ADCC), opsonization, phagocytosis, and neutralization of toxins and viruses (see Table 23-2 ). Different heavy-chain isotypes perform different functions preferentially. IgM, IgG1, IgG2, and IgG3 activate the complement cascade, whereas only IgG1 and IgG3 bind effectively to phagocytic Fcγ receptors. IgE is able to bind to the Fcε receptors expressed on mast cells and eosinophils. In addition, secreted IgA antibodies play a role in protection from invasion through mucosal surfaces.
Different classes of antibodies not only have different functions but also predominate in different compartments in the body. IgA is predominantly secreted, IgM is primarily intravascular, and IgG is generally the major antibody found in tissues and peripheral blood. IgG is able to cross the placenta to provide some protective immunity to the fetus; passive protection from long-lived IgG molecules confers postnatal immunity. However, although the function and location of the antibody classes differ, subpopulations of B cells are able to migrate and home to specific regions of antigen presentation. Therefore generalized B-cell immunity may result from localized antigen presentation, as paradigmatically demonstrated by the efficacy of successful vaccination programs.
T-Cell Activation and Function
CD4+ and CD8+ TCR αβ T cells exported from the thymus circulate in a naïve or resting state in the blood and lymphoid organs but are poised to be activated by cognate antigen presented by MHC on the surface of APCs. CD8+ cytolytic T cells induce the lysis of foreign cells, such as infected, malignant, or allogeneic cells. CD4+ helper T cells interact with B cells via cell-cell and soluble factors to induce class switching and the generation of antigen-specific antibodies. They also provide “help” to CD8+ cytolytic T cells. Activation of resting T cells not only relies on signaling cascades downstream of the TCR-MHC interaction (signal 1) but also requires or is enhanced by the binding of a variety of T-cell surface molecules to their ligands on APCs (signal 2). The lack of a second signal generally results in nonresponsiveness or anergy of the T cell and serves to ensure that activation of the naïve T cell is not only antigen specific but regulated as well. The affinity of the cognate antigen, the engagement of specific costimulatory molecules, and the cytokine milieu collectively influence and modulate the outcome of T-cell activation and thereby result in the generation of distinct effector cell subsets. Although most effector T cells perform their function and expire, a subset of activated cells differentiate into long-lived memory cells capable of rapid anamnestic responses.
T-Cell Receptor Triggering and Activation
The αβTCR is a multimeric transmembrane complex composed of a disulfide-linked antigen-binding clonotypic heterodimer in noncovalent association with the signal-transducing CD3 subunits (CD3εγ, CD3εδ, and CD3ζζ) ( Fig. 23-14 , A ). TCR signaling via CD3 dimers evokes T cell lineage commitment and repertoire selection during development, maintains the peripheral T cell pool, and further differentiates naïve T cells into effector or memory cell populations upon immune stimulation. Each CD3ε, γ, and δ subunit contains an extracellular Ig-like domain, a membrane-proximal stalk region, a transmembrane segment, and a cytoplasmic tail. The interaction between an αβ TCR heterodimer on the T cell and a pMHC ligand on an antigen-presenting cell (APC) initiates a cascade of downstream signaling events. These events are transmitted via the immunoreceptor tyrosine-based activation motif (ITAM) elements in the cytoplasmic tails of the associated CD3 subunits, whose lengths are substantial relative to those of the TCR α and β tails. The various CD3 chains induce distinct patterns of cellular protein tyrosine phosphorylation on activation to recruit intracellular adaptors and signaling molecules. Early, intermediate, and late gene activation programs ensue. Reviews such as Rudolph and colleagues have focused on the structural nature of immune recognition involving Vα and Vβ domains of a given TCR and its pMHC ligand. How recognition of pMHC by a weakly interacting αβ TCR heterodimer on the T cell surface evokes intracellular signaling by way of the adjacent CD3 components of the TCR complex has remained undefined.