Primary short stature—Skeletal abnormalities
With recognizable skeletal dysplasia (achondroplasia, hypochondroplasia)
Without recognizable skeletal dysplasia (Turner syndrome, SHOX gene haploinsufficiency)
Secondary short stature
Malnutrition
Psychosocial deprivation
Chronic diseases
Renal (renal failure, tubular acidosis, nephrotic syndrome)
Intestinal (celiac disease, intestinal inflammatory disease)
Hematological (chronic anemia)
Cardiac
Pulmonary (cystic fibrosis)
Endocrine
Hipothyroidism
Disorders of the GH/IGF-1 axis
Cushing’s syndrome
Pseudohipoparathyroidism
Rickets
Short stature without recognizable cause
Intrauterine growth retardation without recognizable cause (small for gestational age with failure of catch-up growth)
Idiopathic short stature
Familial short stature
Constitutional delay of growth and puberty
Table 16.2
Disorders of the GH/IGF-1 axis
GH deficiency |
Idiopathic |
Acquired (craniopharyngioma, pituitary tumors, autoimmune diseases, granulomatous diseases, central nervous system infections, head trauma) |
Genetic |
GH secretion (GH1 and GHRHR genes) |
Pituitary cells differentiation (POU1F1 and PROP1 genes) |
Pituitary development (HESX1, GLI2, LHX3, LHX4, and SOX3 genes) |
Bioinactive GH |
GH1 gene mutation |
GHInsensitivity |
Primary |
Laron syndrome (GHR gene) |
Associated to immunodisfunction (abnormalities of GH signal transduction, e.g., STAT5B gene defect) |
Secondary or acquired (AntiGH antibodies, malnutrition, liver disorders, diabetes mellitus poorly controlled, uremia) |
Ternary complex formation deficiency (IGF-1/IGFBP-3/ALS) |
Acid-labile subunit deficiency (IGFALS gene) |
IGF-1deficiency |
IGF1 gene |
Bioinactive IGF-1 |
IGF1 gene mutation |
IGF-1 insensitivity |
IGF1R defects and post receptor defects |
Clinical evaluation starts with a detailed medical and family history and a thorough physical examination (Table 16.3). A keypoint is a detailed description of the child’s growth pattern, including the time when the growth deficit was first observed. Birth characteristics must be evaluated (gestation and delivery conditions or complications; gestational age, birth weight, length and head circumference). This information is important to distinguish short children in two groups by the onset of the growth impairment: short stature with prenatal or postnatal onset. Medical history must be investigated, with a focus in neuropsychomotor development, nutritional status, medication use, and cardiac, renal, pulmonary and gastrointestinal diseases. Evaluation of a child’s height must take into account the familial patterns of growth and puberty [1].
Table 16.3
Specific diagnostic findings and keypoints in medical history and physical examination of children with short stature
Findings and keypoints | Interpretation and application |
|---|---|
Medical History | |
Birth length, weight, head circumference, gestational age | Classification as SGA or AGA |
Previous growth data | Height velocity and growth pattern analysis |
Age at start of pubertal signs | Early, normal, or delayed puberty |
Previous diseases, surgeries, and medication use | Organic or iatrogenic causes |
Medical history of the various systems | Search for chronic and systemic diseases |
Feeding and nutrition history | As example, Silver–Russell and Prader–Willi syndromes can lead to feeding difficulties |
Neuropsychomotor development delay and/or intellectual disability | Syndromes, chromosomal disorders, metabolic disorders |
Consanguinity | Likelihood of recessive genetic disorders |
Parental height (measured) | To estimate the target height |
Parents’ age at the start of puberty | To assess likelihood of a familiar pattern of delayed puberty |
Physical Examination | |
Length or height, weight, head circumference, sitting height, arm span, forearm length | Altered sitting height–height ratio is suggestive of skeletal dysplasia |
Weight-for-height or BMI showing underweight | Weight more affected than height, low weight-for-height and low BMI are suggestive of malnutrition |
Weight-for-height or BMI showing overweight or obesity | Hypothyroidism, Cushing’s syndrome, GH deficiency, pseudohypoparathyroidism |
Dysmorphic features | Syndromes |
Pubertal stage | Early, normal or delayed puberty |
General physical exam | Search for chronic and systemic diseases |
Physical examination should be complete, including the description of anthropometric measurements, facial and body dysmorphic features, and any other clues for one of the many causes of short stature. In children younger than 2 years of age, supine length, weight, weight-for-length, and head circumference will be measured, and fontanelles as well as dentition should be evaluated. In older children, erect height, weight, body mass index (BMI), head circumference, arm span, and sitting height (SH) should be measured [1]. In the latter, the pubertal staging has to be evaluated, as determined by Marshall and Tanner criteria [6, 7]. Evaluation of a child’s height must be done in the context of normal standards for sex and age with the international data at hand. Such standards can be either cross-sectional (by calculation of height SDS) or longitudinal (by plotting in growth charts). Serial measurements with a minimum interval of 6 months are necessary to determine the height velocity. Because genetic factors are important determinants of growth and height, all children should be assessed considering siblings and parents. For that purpose, the parental target height is calculated and expressed as mentioned above. When a child’s growth pattern clearly deviates from that of parents and siblings, the possibility of an underlying pathology should be considered [3].
Many abnormal growth states are characterized by disproportionate growth, which is strongly suggestive of skeletal dysplasia. Therefore, body proportion measurements should be part of the evaluation of short stature. We recommend the use of sitting height: height ratio (SH:H) for age and sex, which can also be expressed in SDS, according to published standards. This ratio allows for the observation of body proportion changes throughout development. Children with short stature and an increased SH:H ratio for age and sex have a disproportional short stature caused by limb abnormalities, while children with short stature and a decreased SH:H ratio for age and sex have a disproportional short stature caused by axial segment abnormalities [1, 8] (Fig. 16.1).
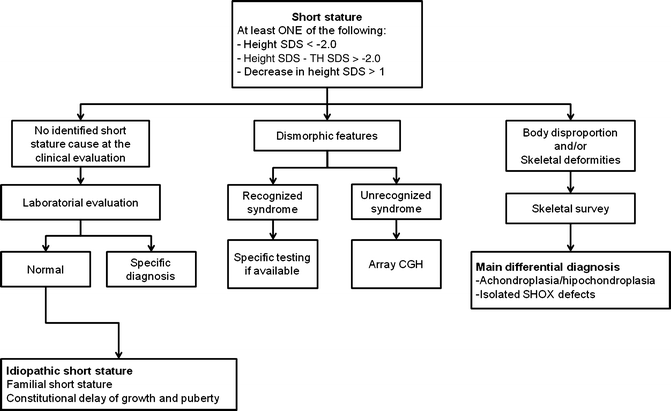
Fig. 16.1




Diagnostic approach in children with short stature. SDS = standard deviation score; TH = target height; CGH = comparative genomic hybridization
Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
