HYPOGONADOTROPIC HYPOGONADISM
The term hypogonadotropic hypogonadism (Table 92-3) applies to a variety of diagnoses, all united by the feature of ineffective gonadotropin production. It can result either from a true deficiency of gonadotropins due to primary disease in the hypothalamus or pituitary (primary hypothalamic/pituitary insufficiency) or as a consequence of a reversible gonadotropin deficiency that is due to another illness or condition (secondary hypothalamic/pituitary insufficiency).
 |
CONSTITUTIONAL DELAY
The majority of children with pubertal delay will have constitutional delay of puberty (CDP) (Table 92-4), a transient functional delay in sexual development that is a variant of normal. Although these adolescents ultimately progress through puberty normally, pubertal onset is sufficiently late to trigger concern and evaluation. CDP can occur either as an isolated finding, or, more commonly, it may be associated with concomitant short stature (constitutional delay of growth and puberty, or CDGP). In fact, many of these children initially come to medical attention because of height concerns, not pubertal delay. In either case, there often is a positive family history of similar developmental patterns, and physical examination and laboratory testing show the child to be without underlying disease.
 |
Children with constitutional delay have a temporary delay in the maturation of the hypothalamic–pituitary–gonadal axis and a protracted phase of prepubertal growth deceleration (Fig. 92-5). The exact etiology is not clear. Transient GH deficiency, relative to the chronologic age, is well described in children with constitutional delay. Whether low insulin-like growth factorI (IGF-I) levels impair the gonadal response to gonadotropins76 or whether GH deficiency is instead a consequence of the low sex steroid levels77 is unknown. However, since GH secretion normalizes after the onset of puberty in most cases,78 the latter seems more likely. Deficiency of the transcription factor, Otx1, has been implicated in transient hypogonadotropic hypogonadism and GH deficiency in mice79; this insight may someday lead to the identification of the genetic basis for constitutional delay in humans.
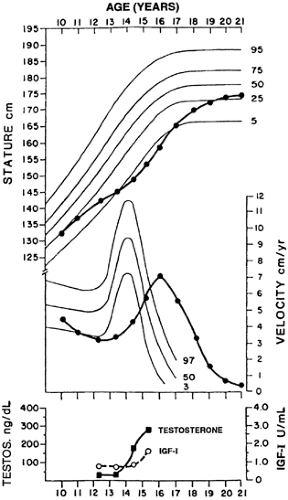 FIGURE 92-5. Growth pattern and hormonal changes in a boy with constitutional delay of growth and puberty. He ultimately entered puberty and achieved normal adult height, despite a delayed growth spurt. Testosterone and insulin-like growth factor-I (IGF-I) belatedly rose to normallevels. (Modified from Rosenfield RL. Clinical review 6: diagnosis and management of delayed puberty. J Clin Endocrinol Metab 1990; 70:559.) |
The prototypic adolescent with CDP is a boy, possibly short all of his life, who now seeks help for a delay in the onset of secondary sexual characteristics. Bone age is invariably delayed. In most cases, the growth velocity is within two SD of normal, especially for the bone age, and the height is also appropriate for bone age. Eventually, normal puberty will ensue before the bone age is 13 years. Children with CDGP should achieve a normal adult height, but there is controversy regarding whether these children achieve their genetic potential. Some studies demonstrate that final height is not significantly different from midparental height,80 whereas others suggest that the target height is not achieved.81
Many more boys than girls are evaluated for delayed puberty. This may reflect the natural genetic and physiologic differences between boys and girls or, more likely, a societal bias regarding growth and development. Boys, particularly those with concomitant short stature, are more prone to teasing and stigmatization from a delay in pubertal development. The social repercussions and resultant psychological effects of constitutional delay can be quite severe; therefore, it is important that these issues always be thoughtfully addressed in these children.
PRIMARY HYPOTHALAMIC/PITUITARY INSUFFICIENCY
Gonadotropin deficiency may occur either as an isolated event or as a component of multiple pituitary hormone deficiencies or hypothalamic dysfunction (Fig. 92-6). Essentially, any event or condition that results in hypopituitarism can cause hypogon-adotropic hypogonadism. Predisposing factors include head trauma, infection, CNS irradiation, and intrinsic developmental or anatomic defects such as septooptic dysplasia (de Morsier syndrome) or primary empty sella (Fig. 92-7). Most of these aforementioned problems are diagnosed before adolescence, since these children usually present earlier with associated endocrine deficiencies or growth failure. On the other hand, isolated gonadotropin deficiency may not be diagnosed until adolescence when pubertal delay is the primary complaint.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


