Introduction
One of the most important metabolic events to mark the transition from fetal to neonatal life is the adaptation from an environment that has a readily available and continuous source of glucose—transplacental passage of maternal blood—to an environment in which glucose is provided in a limited and intermittent supply via feeding. At birth, after separation from the placenta, the complex mechanisms involved in the maintenance of plasma glucose concentration must be activated and coordinated to avoid hypoglycemia and resultant damage to the central nervous system. A newborn or infant with hypoglycemia presents an urgent diagnostic and therapeutic challenge. The clinical features must be rapidly assessed and a plan of action developed based on the infant’s age, maternal and parturition history, severity and persistence of hypoglycemia, and all other relevant clinical clues for definitive diagnosis and treatment. In this chapter, we review the normal physiology of glucose metabolism and the pathophysiology of the most common disorders resulting in hypoglycemia in the neonate (first 4 weeks of life) and infant (1 month to 1 year of life) with an emphasis on the approach to the diagnosis and management of these pathologies.
Principles of glucose metabolism
A systematic approach to hypoglycemia in the newborn, infant, or child requires an appreciation of the central role of glucose in the body’s fuel economy. Glucose metabolism accounts for approximately half of basal daily energy needs and is the principal metabolic fuel of the human brain. Glucose can be stored for energy in the form of glycogen and fat, although only glucose stored in liver glycogen is available for release into the circulation, and fatty acids cannot be converted back into glucose; glucose carbon can be used for synthesis of protein and for structural components (such as cell membranes). The aerobic oxidation of glucose yields high energy by producing 36 mols of adenosine triphosphate (ATP) for each mol of glucose. Although glucose is the predominant metabolic fuel for both the brain and the body in the period immediately after a meal, during longer term fasting, glucose utilization must be restricted to glycolytic tissues, such as red blood cells and the brain and the rest of the body must rely on oxidation of fatty acids released from adipose tissue triglyceride stores.
All glucose extracted by the brain is oxidized, and thus cerebral glucose utilization parallels cerebral oxygen uptake. In 5-week-old infants, cerebral glucose utilization already represents 71% to 93% of the adult level in most brain regions (ranging from 13 to 25 μmol/100 g/min). Adult levels of cerebral glucose utilization (19–33 μmol/100 g/min) are reached by 2 years of age, and they continue to increase until 3 to 4 years of age—when they reach values ranging from 49 to 65 μmol/100 g/min, which are maintained to approximately 9 years of age and then gradually decline to adult levels by the end of the second decade.
Glucose uptake by the brain occurs by means of a carrier-mediated facilitated diffusion process that is glucose concentration dependent, as well as energy, Na + , and insulin independent. This process is mediated by facilitative glucose transporter (GLUT) proteins. Several members of the GLUT family have been detected in the brain. GLUT1 is located at the blood-brain barrier, and although some neurons express GLUT2 and GLUT4, the majority use GLUT3 as their primary transporter. The main isoform present in insulin-responsive tissues is GLUT4.
Of major importance is that glucose entry into brain cells and its subsequent metabolism are not dependent on insulin but rather are dependent on circulating arterial plasma glucose concentration. Therefore a decrease in arterial glucose concentration or a defect in the glucose transport mechanism of the brain will result in intracerebral glucopenia and low cerebrospinal fluid glucose concentration (hypoglycorrhachia)—with attendant symptoms and signs of cerebral glucopenia as subsequently described. For example, genetic deficiency of the GLUT1 glucose transporter causes hypoglycorrhachia, seizures, and severe developmental delay.
In normal adult humans and in the normal newborn infant after the first few days of life, plasma glucose concentrations range from 3.9 to 5.6 mmol/L (70–100 mg/dL). In experiments using rats, brain glucose consumption outstrips glucose transport, leading to a brain glucose concentration near zero at a plasma glucose concentration of 2 mmol/L (36 mg/dL). To prevent circulating arterial plasma glucose from decreasing precipitously under normal physiologic conditions, and therefore to prevent impairment of vital function that depends on cerebral glucose metabolism, an elaborate defense mechanism has evolved. This defense against hypoglycemia is integrated by the autonomic nervous system and by hormones that act to enhance glucose production through enzymatic modulation of glycogenolysis and gluconeogenesis, while simultaneously limiting peripheral glucose use. Similar defense mechanisms against hypoglycemia function in the normal full term human newborn after separation from the placenta. Thus hypoglycemia is the result of a defect in one of these systems that maintain a normal range of plasma glucose concentration, preventing its fall to less than 3.9 mmol/L (70 mg/dL) during fasting and its rise to more than 7.8 mmol/L (140 mg/dL) during feeding.
These mechanisms, most importantly, the regulation of β-cell insulin secretion, are not fully mature at birth, when there is an abrupt transition from intrauterine life to extrauterine life, and mean plasma glucose concentrations transiently fall by 25% before recovering by 2 days of life to normal adult values. Neonates who are born with various risk factors, including birth asphyxia, intrauterine growth restriction (IUGR), maternal toxemia, and prematurity may be particularly vulnerable to hypoglycemia, often with consequences to subsequent cerebral development or function that can be prevented by early detection and treatment.
Physiology of perinatal glucose homeostasis
Glucose Metabolism in the Fetus
Glucose provides 80% of fetal energy, with the remaining 20% derived mostly from oxidation of lactate and amino acids. Free fatty acids are transported across the placenta in limited amounts sufficient for fetal growth, but do not contribute significantly to fetal energy demands. The glucose used by the fetus is derived entirely from the maternal circulation through facilitated diffusion across the placenta using primarily the GLUT1 glucose transporter. Placental glucose transport does not involve the GLUT4 glucose transporter, and therefore is not regulated by either maternal or fetal insulin concentrations. Because placental glucose transport is so rapid, the maternal and fetal glucose concentrations behave as a single pool and there is no endogenous glucose production in the fetus. This has important clinical consequences, for example, acute hypoglycemia in a mother will result in acute hypoglycemia in the fetus—with little or no ability of the fetus to acutely compensate for the abrupt reduction in glucose supply.
From early gestation to the time of delivery, maternal arterial and fetal umbilical vein concentrations of plasma glucose are very similar. The mean maternal-fetal plasma glucose concentration difference is essentially zero at 20 weeks of gestation and increases only slightly to 0.75 mmol/L (13 mg/dL) at term. The small increase in maternal-fetal gradient may reflect increased utilization by the growing placenta, because approximately 50% to 60% of maternal glucose taken up is consumed by oxidation in the placenta and only 40% to 50% transported to the fetus. As gestation progresses, the increasing demand of the fetus for glucose oxidation and storage is accomplished by increasing uterine blood flow. There is excess capacity in uterine blood flow, and it may be reduced as much as 50% without causing any fetal harm. However, reducing blood flow from the placenta to the fetus causes significant impact on delivery of glucose and other nutrients to the fetus, with resultant IUGR.
In the second half of pregnancy, the fetal-placental unit secretes large quantities of placental lactogen, progesterone, and estrogen, which cause increased maternal insulin resistance and may, in susceptible mothers, result in gestational diabetes. Maternal insulin secretion is responsible for controlling plasma glucose concentrations in the mother and, while insulin does not cross the placenta (unless bound by antibodies), the maternal insulin also controls fetal glucose concentrations, because of the rapid facilitated transfer of glucose across the placenta. Although fetal insulin does not control fetal glucose concentrations, fetal insulin secretion is responsive to changes in fetal glucose concentrations. The role of fetal insulin is to maintain the rapid growth rate of the fetus. For example, maternal hyperglycemia (e.g., maternal diabetes) leads to fetal hyperinsulinemia and increased fetal growth; in contrast, maternal hypoglycemia (as in maternal malnutrition or in maternal hyperinsulinism caused by a glucokinase activating mutation) can suppress fetal insulin secretion, resulting in impaired fetal growth.
Changes at Birth: Transition Phase
The abrupt interruption of maternal glucose transfer to the fetus at delivery imposes a need for the newborn infant to independently control plasma glucose concentrations by adjusting insulin secretion and mobilizing counterregulatory responses. There is evidence that most of the counterregulatory mechanisms, including glucagon and epinephrine secretion and activation of hepatic glycogenolysis and gluconeogenesis, are functional at birth. Nevertheless, there is a transitional period immediately after delivery when mean plasma glucose concentrations fall in normal newborn infants from 70 to 80 mg/dL (close to maternal glucose values) to 55 to 60 mg/dL before returning by 2 to 3 days of life to the range of 70 to 100 mg/dL, which is normal for extrauterine life. This phenomenon, known as transitional neonatal hypoglycemia , in normal newborns has been recognized for many years, although its clinical significance remains uncertain, indeed controversial. There is increasing evidence that the mechanism of transitional hypoglycemia involves pancreatic regulation of insulin secretion and reflects persistence of the lower glucose threshold for β-cell insulin secretion in the fetus, which is required for maintaining fetal growth. Evidence that transitional neonatal hypoglycemia should be considered as “transitional neonatal hyperinsulinism” includes observations that, during the period that plasma glucose is low in normal newborns, lipolysis and ketogenesis are suppressed and liver glycogen reserves are maintained, as shown by the large glycemic responses elicited by administration of glucagon or epinephrine. As discussed later in this chapter, these findings are diagnostic hallmarks of insulin-induced hypoglycemia. The lower concentrations of plasma glucose in normal newborns are quite stable and relatively unaffected by durations of postnatal fasting of up to 24 hours, suggesting that transitional neonatal hypoglycemia reflects a lower glucose threshold for insulin secretion. Recent studies in mice confirm a lower glucose threshold for insulin secretion in the immediate postnatal period. The mechanism responsible for the low glucose threshold in fetal beta-cells remains unclear, but suggested explanations include expression of genes that are normally “disallowed” in the beta-cell, such as hexokinase-1, the plasma membrane transporter for pyruvate and lactate, and lactate dehydrogenase or heightened sensitivity of insulin granule release to low glucose. The phenomenon of transitional neonatal hyperinsulinism in normal newborns appears to usually be benign, but often complicates the recognition of infants who have persistent, pathological hypoglycemia disorders (see later).
Other challenges to the regulation of plasma glucose concentrations after delivery include the need to switch from a constant supply of glucose and amino acids provided from the maternal circulation to a variable and intermittent oral intake of nutrients. In breastfed infants, the composition of feedings also changes over the first days of life, with colostrum being lower in carbohydrates and higher in fat than mature breast milk. Breastfed babies frequently may not achieve full nourishment initially and, beyond the period of transitional neonatal hyperinsulinism, may develop mild fasting hyperketonemia. Although this hyperketonemia was previously often attributed to the high fat content of colostrum (“suckling ketosis”), it appears to reflect a response to mild fasting stress. Impairments in adaptation to these changes in fuel supply in newborns may be manifested as symptomatic hypoglycemia, as described in later sections on specific hypoglycemia disorders in neonates and early infancy. Insulin secretion is glucose-responsive during late fetal development, but maximum capacity may be limited, especially in extremely premature infants who require intravenous (IV) feedings.
In summary, plasma glucose concentrations during intrauterine life are constant and are maintained to greater than 3.9 mmol/L (70 mg/dL). Following birth, plasma glucose falls to a mean glucose of 3.1 to 3.3 mmol/L (56–60 mg/dL), during the first 12 hours as a consequence of the low beta-cell glucose threshold for insulin secretion. As insulin regulation adapts to extrauterine life, glucose concentration rises to a mean of 3.5 mmol/L (63 mg/dL) by 12 to 24 hours of life; by 48 to 72 hours plasma glucose rises to a mean of 4.1 mmol/L (74 mg/dL). The distribution of plasma glucose concentrations in neonates immediately after birth is not “normal” but is skewed toward lower values because of babies with various risk factors for hypoglycemia, as described later. For this reason, approximately 30% of “normal” newborns will have plasma glucose concentrations lower than 2.8 mmol/L (50 mg/dL) in the first 8 to 12 hours of life, but thereafter the frequency of glucose concentrations greater than 50 mg/dL drops to only 0.5% in newborns older than 24 hours of age. This drop in plasma glucose levels in the first 24 hours of life is transitional hypoglycemia (transitional neonatal hyperinsulinism) , which by definition occurs in normal healthy newborns. It should be differentiated from both transient and persistent pathologic causes of hypoglycemia.
Abnormalities of Transition
Studies of plasma glucose concentrations immediately after delivery in normal newborns show not only a lower mean concentration of glucose, but also a wider variance and, as noted earlier, a skewing of the distribution toward glucose values. This means that the simple mean ± 2 standard deviations (SDs) is not a valid measure of the normal range of glucose in newborns, although this is used as the basis for many guidelines on neonatal hypoglycemia. The excess of low glucose concentrations reflects the fact that a large proportion of neonates are at increased risk of potentially pathologic hypoglycemia during the period of transitional neonatal hyperinsulinism. These at-risk infants have more severe and more prolonged hypoglycemia after delivery, which is caused by pancreatic insulin secretion and has been termed perinatal stress-induced hyperinsulinism . Perinatal stress-induced hyperinsulinism occurs with a number of fetal and maternal disorders, such as IUGR, birth asphyxia, maternal diabetes, and maternal hypertension ( Box 7.1 ). Whether perinatal stress-induced hyperinsulinism is simply an exaggerated and more protracted form of transitional neonatal hyperinsulinism is not known. Affected neonates may require treatment to control hypoglycemia for several weeks or months after birth. Premature infants may not have developed adequate hepatic glycogen stores and may also have immaturities of liver enzyme systems for gluconeogenesis and ketone synthesis, which increase their risk of hypoglycemia but to a lesser extent than in infants with perinatal stress. It is important for the clinician to recognize these at-risk neonates and neonates whose glucose control is out of the range of transitional hypoglycemia during this period and treat appropriately. In particular, it is important to recognize conditions in which insulin secretion is increased, as these infants will neither mobilize glycogen nor be able to oxidize fatty acids, thus putting their brains at risk of damage because of the combination of hypoglycemia and hypoketonemia.
Infants of Diabetic Mothers (including gestational diabetes)
Large for Gestational Age (LGA) (even without maternal diabetes)
Perinatal Stress-induced Hyperinsulinism
Intrauterine Growth Restriction (IUGR), Small for Gestational Age (SGA)
Birth Asphyxia; C-section for fetal distress
Maternal preeclampsia or hypertension
Meconium aspiration syndrome
Erythroblastosis fetalis
Polycythemia
Premature or Postmature Delivery
Family History of a Genetic Form of Hypoglycemia
Congenital Syndromes Associated With Hypoglycemia
Beckwith-Wiedemann Syndrome (especially 11pUPD)
Turner Syndrome
Kabuki Syndrome
Midline facial malformations, microphallus
Management of Hypoglycemia in the First 24 to 48 Hours
Management of hypoglycemia in the first 24 hours of life is important from two points of view. First, the management plan should avoid treatment of infants who do not need treatment. Second, it should include a plan for those who either have or are at risk of having a persistent hypoglycemic disorder, which both identifies those neonates and treats them adequately. It is not currently recommended that all newborns have plasma glucose concentrations measured. However, if an infant has symptoms consistent with hypoglycemia—such as lethargy, apnea, or seizures, or is unwell or in any of the at-risk categories (e.g., infants with siblings with known hypoglycemic disorders, preterm infants, large for gestational age infants, small for gestational age infants, infants of a diabetic mother, infants who had birth asphyxia, and other infants with disorders outlined in Box 7.1 )—the infant’s glucose levels should be measured.
The recent recommendations of the Pediatric Endocrine Society suggest that beyond the period of normal transitional neonatal hypoglycemia in the first 24 to 48 hours after birth, the glucose thresholds and goals for management of low plasma glucose concentrations should be the same in neonates as in older children. In addition, these same standards are recommended for neonates who either have or are at increased risk of having a persistent hypoglycemia disorder (e.g., family history or physical features of a hypoglycemia disorder, such as large for gestational age birthweight, suggesting hyperinsulinism or macroglossia, and hemihypertrophy, suggesting Beckwith-Wiedemann syndrome [BWS]). For neonates who are suspected to not have a persistent disorder and who are receiving normal feedings, the recommended glucose target for management is to maintain plasma glucose above 50 mg/dL during the first 48 hours of life and above 60 mg/dL beyond 48 hours, if the hypoglycemia is likely to resolve within a day or two.
Emergency treatment of hypoglycemia in neonates should be tailored to the presence or absence of symptoms and the degree of hypoglycemia. For mild asymptomatic hypoglycemia in the first 48 hours of life, early feeding or oral glucose may be tried initially. However, symptomatic or severe hypoglycemia should be treated with IV dextrose (e.g., 0.2 g/kg bolus, followed by a glucose infusion rate [GIR] of at least 4–6 mg/kg/min initially, and adjusted as needed to maintain glucose > 70 mg/dL). If the hypoglycemia is known to be caused by hyperinsulinism, glucagon may also be considered (0.5–1 mg IV, subcutaneous [SQ], or intramuscular [IM]). Follow-up frequent monitoring of plasma glucose should be done to determine whether formal diagnostic evaluation is required.
By 48 hours of age, the period of transitional neonatal hypoglycemia should normally be over and plasma glucose concentrations should be similar to older children and adults: a normal range of 70 to 100 mg/dL. Thus, formal evaluation to diagnose the cause of hypoglycemia should be considered after 48 hours of age for neonates in whom the possibility of having a persistent hypoglycemia disorder cannot be completely excluded. Such an evaluation to diagnose the cause of hypoglycemia should be undertaken before the baby is allowed to be discharged home. To address the issue of babies who may not have achieved complete resolution of transitional hypoglycemia before the time of discharge, the Pediatric Endocrine Society recommendations suggest considering a fasting challenge to ensure that plasma glucose can be safely maintained above 60 mg/dL for over 6 to 8 hours (the “skip a feed” test). This should only be considered for neonates who have no risk features for a persistent hypoglycemia disorder (i.e., no episodes of symptomatic hypoglycemia, no need for IV dextrose, no features to suggest genetic or syndromic form of hypoglycemia, or perinatal stress-induced hypoglycemia, etc.). For all other neonates, a complete formal fasting study should be considered. One should note that there is no evidence that proves these protocols recommended by the Pediatric Endocrine Society will identify 100% of infants, so caution should be made if clinical circumstances warrant further thought. Finally, it is important to emphasize to anxious parents the consequences of missing a diagnosis of a hypoglycemic disorder. In our experience, most parents are willing to lengthen the hospital stay by 6 to 9 hours to ensure the long-term safety of their child.
Hormonal and metabolic systems of fasting adaptation
Hypoglycemia in neonates, infants, and children is essentially always a problem with fasting adaptation. Postprandial hypoglycemia is exceedingly rare and is limited to a few unusual situations, such as postprandial hypoglycemia after Nissen fundoplication, hereditary fructose intolerance, or the protein-induced hypoglycemia seen in some forms of congenital hyperinsulinism. Therefore a consideration of the four major hormonal and metabolic pathways that maintain fuel homeostasis during fasting provides an important framework for understanding the causes, diagnosis, and treatment of different forms of hypoglycemia.
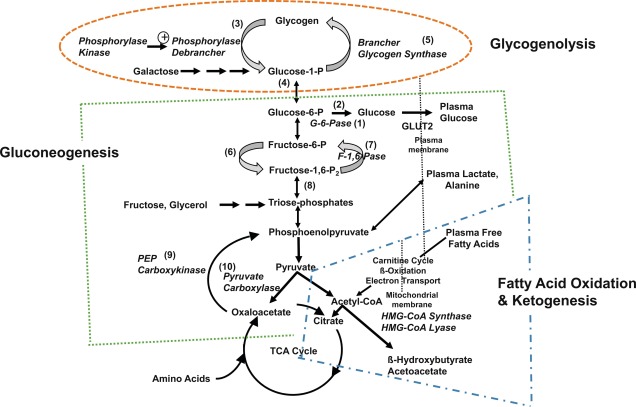
Three metabolic systems regulate the physiologic response to fasting: (1) hepatic glycogenolysis, (2) hepatic gluconeogenesis, and (3) hepatic ketogenesis. The key enzymatic steps in these pathways are shown in Fig. 7.1 . These metabolic systems are coordinated by (4) the endocrine system, consisting of suppression of insulin (the most important endocrine response to fasting, as insulin suppresses all three metabolic systems), and the secretion of the counterregulatory hormones: glucagon, epinephrine (a marker of sympathetic nervous system activation), cortisol, and growth hormone (GH). Table 7.1 summarizes the counterbalancing effects of these counterregulatory hormones on the three key metabolic systems. There is a hierarchic redundancy in the interaction of the counterregulatory hormones that provides a margin of safety (“fail-safe mechanism”) if only one counterregulatory hormone is impaired. Epinephrine (sympathetic activation) and glucagon are quick acting, each signaling its effects by stimulation of cyclic adenosine monophosphate (AMP). Deficiencies of glucagon, as occurs in long-standing type 1 diabetes mellitus, can be largely compensated for by an intact autonomic nervous system with appropriate α- and β-adrenergic, and cholinergic effects. Conversely, autonomic failure can be largely compensated for if glucagon secretion remains intact.
| Counterregulatory Hormone | Glycogenolysis | Gluconeogenesis | Lipolysis | Ketogenesis |
|---|---|---|---|---|
| Insulin | Inhibits | Inhibits | Inhibits | Inhibits |
| Glucagon | Stimulates | Stimulates | Stimulates | |
| Cortisol | Stimulates | |||
| Growth hormone | Stimulates | |||
| Epinephrine | Stimulates | Stimulates | Stimulates |
Hepatic glycogenolysis is sufficient to meet energy requirements for only a few hours. Beyond that time, glucose must be produced by hepatic gluconeogenesis from precursors, such as amino acids, glycerol, and lactate recycled from glycolysis. The major source of gluconeogenic precursors is amino acids from muscle protein. Although the pool of muscle protein is large, it is required for body function and thus in contrast to stores of glycogen in liver and fat in adipose tissue, there are no “reserves” of protein to draw on during fasting. To spare the use of essential protein during extended fasting, glucose consumption must be suppressed by switching on the mobilization of fatty acids from adipose triglyceride stores for oxidation in muscle and other tissues and of glycerol as substrate for hepatic gluconeogenesis. Fatty acids are also oxidized in liver to produce the ketone bodies, β-hydroxybutyrate and acetoacetate, which can be used by the brain as an alternative substrate to further spare glucose consumption.
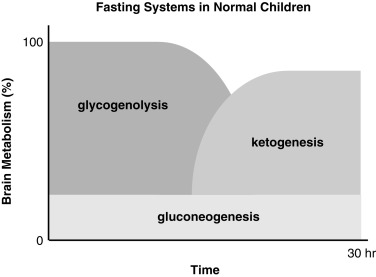
The essential function of fasting adaptation is to maintain fuel supply to the brain. Glucose homeostasis is very limited in neonates and infants compared with adults, in part because of their smaller reserves of liver glycogen and muscle protein, but chiefly because of their relatively larger rates of glucose consumption because of their larger brain-to-body-mass ratio. For example, the fuel stores of a 10-kg infant are only 15% of those of an adult. However, the caloric needs are 60% of those of an adult and glucose utilization rates per kilogram body weight are 2- to 3-fold greater. As shown in Fig. 7.2 , early in fasting after absorption of a meal is completed, glucose is the primary brain fuel and accounts for over 90% of total body oxygen consumption. Glucose is initially provided chiefly from hepatic glycogenolysis, supplemented by hepatic gluconeogenesis using amino acids released by muscle protein turnover and lactate from glycolytic tissues, such as red blood cells. After 8 to 12 hours in normal infants (24–36 hours in adults), glucose production declines, because the supply of liver glycogen is depleted and the rate of gluconeogenesis from amino acids remains constant. At this time, a transition to fat as the major fuel for the body begins, with accelerated adipose tissue lipolysis and increased fatty acid oxidation in muscle and ketogenesis in liver. Lipolysis also generates glycerol, which becomes an important gluconeogenic substrate once fasting adaptation is fully active. The brain cannot directly use fatty acids, because they do not pass the blood-brain barrier. However, the brain can substitute glucose consumption with the ketones acetoacetate and β-hydroxybutyrate, which are released by the liver as the end product of hepatic fatty acid oxidation. In late stages of fasting adaptation, fatty acid oxidation and ketone utilization account for 90% of total body oxygen consumption.
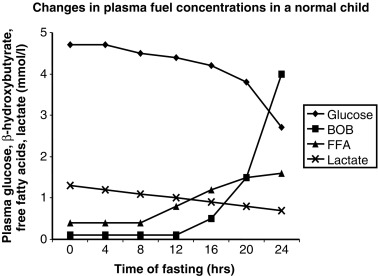
The operation of the metabolic and endocrine systems of fasting adaptation is evidenced by the changes in circulating levels of metabolic fuels and hormones during a fast. As shown in Fig. 7.3 , in young infants, a 24-hour fast is accompanied by a gradual fall in plasma glucose levels as hepatic glycogen stores are depleted, a progressive decline in concentrations of gluconeogenic substrate (e.g., lactate, alanine) as they are used for hepatic gluconeogenesis, a brisk rise in free fatty acids as lipolysis is activated, and a dramatic rise in β-hydroxybutyrate (the major ketone) as hepatic ketogenesis is turned on.
By the time that plasma glucose concentration has fallen to less than 2.8 mmol/L (50 mg/dL), all the metabolic systems and the hormonal responses described earlier will have fully engaged. A “critical sample” drawn at this time will therefore provide a snapshot of the integrity of the four metabolic and endocrine fasting systems ( Table 7.2 ). The use of these critical samples in diagnosing the cause of hypoglycemia is discussed later.
| Disorder | Plasma Fuels at End of Fast (mmol/L) | Plasma Hormones at End of Fast | Physical Examination | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Glucose | Lactate | Free fatty acids | ß-hydroxy- butyrate | Insulin (μu/mL) | Cortisol (ug/dL) | Growth hormone (ng/mL) | Glycemic response to glucagon (mg/dL) | ||
| Normal Infants | 2.8 | 0.7–1.5 | 1.5–2.5 | 2–4 | < 2 | > 20 | > 10 | < 30 | |
| Hyperinsulinism | 2.8 | N | < 1.7 | < 1.8 | > 1 | N | N | > 30 | LGA |
| Cortisol deficiency | 2.8 | N | N | N | N | Low | Low | N | |
| GH deficiency | 2.8 | N | N | N | N | N | Low | N | Short stature |
| Panhypopituitarism | 2.8 | N | N | N | N | Low | Low | N | Short stature, midline facial malformation, optic hypoplasia, micropenis |
| Epinephrine deficiency (beta-blocker) | 2.8 | N | < 1.5 | < 2 | N | N | N | N | |
| Debrancher deficiency (GSD III) | 2.8 | N | N | N | N | N | N | N | Hepatomegaly 4 + |
| Phosphorylase deficiency (GSD VI) | 2.8 | N | N | N | N | N | N | N | Hepatomegaly 2 + |
| Phosphorylase kinase deficiency (GSD IX) | 2.8 | N | N | N | N | N | N | N | Hepatomegaly 2 + |
| Glycogen synthase deficiency (GSD 0) | 2.8 | N | N | N | N | N | N | N | Hepatomegaly 1 + |
| Glucose 6-phosphatase deficiency (GSD Ia and Ib) | 2.8 | 4–8 + | N | < 2 | N | N | N | (lactate) | Hepatomegaly 4 + |
| Fructose 1,6-diphosphatase deficiency | 2.8 | 4–8 + | N | N | N | N | N | N | Hepatomegaly 1 + |
| Pyruvate carboxylase deficiency | 2.8 | 4-8 + | N | N | N | N | N | N | |
| Lipolysis | |||||||||
| Congenital lipodystrophy, familial dysautonomia, beta-blockers | 2.8 | N | < 1.5 | < 2 | N | N | N | N | |
| Fatty acid oxidation defect | 2.8 | N | > 2.5 | < 1.5 | N | N | N | N | |
Definition hypoglycemia in neonates and infants
The definition of hypoglycemia is an important concept to assist in diagnosis and management of patients with suspected or proven hypoglycemic disorders. For this reason, we recommend using various different plasma glucose thresholds for different definitions. The diagnostic threshold is the level of glucose below or at which a diagnosis of the etiology of hypoglycemia may be made. The therapeutic threshold is the level of glucose that indicates adequate therapy of a disorder of glucose dysregulation. Finally, there is the very controversial issue of what level of glucose causes brain damage.
A number of potential artifacts can interfere with measuring glucose levels in neonates and infants ( Box 7.2 ). Whole-blood glucose concentrations are 10% to 15% lower than plasma glucose concentrations because erythrocytes have a higher concentration of protein (hemoglobin) versus plasma with its higher water content, and hence higher dissolved glucose concentrations. The difference may be greater in neonates with higher hematocrits. Blood samples that are not processed promptly can have erroneously low glucose levels, owing to glycolysis by red and white blood cells. At room temperature, the decline of whole-blood glucose can be 5 to 7 mg/dL/h. The use of inhibitors, such as fluoride, in collection tubes avoids this problem.
- •
Whole-blood versus plasma glucose concentration (plasma is 10%–15% higher)
- •
Duration between sample collection and sample measurement
- •
Presence or absence of glycolytic inhibitors in collection tubes
- •
Sample collection from indwelling lines without adequate flushing
Hospital bedside glucose meters and similar home glucose meters are less precise than clinical laboratory methods and can be expected to have an error range of 10% to 15%. These methods are also prone to errors, such as outdated strips or inadequate sampling—most of which result in falsely low glucose values. For this reason, bedside monitors can be used for screening purposes—but any glucose value below 3.3 mmol/L (60 mg/dL) should be verified in the clinical laboratory. Falsely low (or high) glucose values may occur with samples drawn from indwelling lines without adequate flushing of the saline (or glucose) infusate. Falsely low levels may also occur in critically ill infants with poor perfusion and use of bedside meters in the critical care setting is recommended only for venous or arterial samples. Modern bedside monitors use whole-blood samples but generally are calibrated to give a result expressed as a plasma glucose.
Diagnostic Glucose Threshold
The classic definition of symptomatic hypoglycemia is Whipple’s triad—symptoms, signs or both consistent with hypoglycemia, a low plasma glucose concentration, and resolution of symptoms/signs after the plasma glucose concentration is raised. These three criteria were originally used for diagnosing insulinomas in adults. However, in neonates and infants, who cannot dependably communicate their symptoms and in whom clinical signs of hypoglycemia are not specific, it may not be possible to satisfy Whipple’s triad. In these cases, recognition of hypoglycemia may require confirmation by repeated measurements of plasma glucose concentrations, either in association with symptoms or signs of hypoglycemia or during screening of at-risk infants. If levels are found below the level of glucose that causes neurogenic symptoms (< 3.3 mmol/L or < 60 mg/dL), after the first 48 hours of life, further investigations should be carried out to determine the etiology. In these instances, formal provocative testing should be performed to determine if the neonate is safe for discharge or to determine the etiology of hypoglycemia.
Beyond the first 48 hours of life, normal plasma glucose concentration in neonates and infants are not different than in older children and adults. Thus plasma glucose concentrations in the postabsorptive state range between 3.9 and 5.6 mmol/L (70 and 100 mg/dL), with a mean of 4.4 to 4.7 mmol/L (80–85 mg/dL). A plasma glucose level of 2.8 mmol/L (50 mg/dL) is conventionally used as an end point for provocative diagnostic tests for hypoglycemia. This value is low enough to strongly stimulate the endocrine and metabolic defenses against hypoglycemia for identifying the mechanism responsible for hypoglycemia. Falling glucose levels elicit a typical sequence of responses: plasma insulin levels begin to decrease when plasma glucose falls to the range of 4.4 to 4.7 mmol/L (80–85 mg/dL) and insulin secretion is generally “switched off” at glucose concentrations below 2.5 to 3 mmol/L (45–54 mg/dL); glucagon secretion increases when plasma glucose levels are in the range of 3.6 to 3.9 mmol/L (65–70 mg/dL); epinephrine, cortisol, and GH responses are activated in the range of 3.6 to 3.9 mmol/L (65–70 mg/dL). As the plasma glucose falls below 3.3 mmol/L (59 mg/dL), auditory and visual reaction time is prolonged and cognitive function begins to decline as the plasma glucose concentration declines below 2.5 to 3.5 mmol/L (45–63 mg/dL), with some of this variability dependent of the testing used.
Therapeutic Threshold
Once a diagnosis of the etiology of hypoglycemia has been made or suspected, the goal of therapy is to maintain the plasma glucose concentration > 3.8 mmol/L (> 70 mg/dL). This target allows patients with hypoglycemia disorders to maintain glucose levels above the threshold for counterregulatory responses and prevent the development of hypoglycemia unawareness caused by repetitive hypoglycemia. For disorders of fatty acid oxidation and gluconeogenesis, maintaining glucose concentration in this range will prevent buildup of free fatty acids and lactate, respectively.
Plasma glucose concentrations between 2.8 and 3.9 mmol/L (50 and 70 mg/dL) should be regarded as suboptimal and below the goal for therapy for hypoglycemia and should lead one to evaluate if a therapy escalation is needed.
Hypoglycemic Brain Damage
“What level of glucose causes brain damage?” is one of the most frequently asked questions. Unfortunately, the answer is more complex than the lower the glucose, and the longer the glucose is low, the worse the risk. Brain damage likely occurs because of fuel deficiency in neurons. Neurons have many sources of energy, the most common of which are glucose, lactate, and β-hydroxybutyrate. Studies of normal newborn infants have shown that after 6 hours of fasting, ketones can account for 12% of energy production by the brain.
To fully oxidize these compounds to energy, the cell needs oxygen and adequate blood flow. Indeed, one of the early brain adjustments to hypoglycemia (at approximately 30 mg/dL) is to increase cerebral blood flow delivering more glucose and oxygen to the brain. Studies have shown that the addition of hypoxemia, asphyxia, or ischemia to hypoglycemia dramatically increase the extent of brain damage compared with hypoglycemia alone, and the presence of hypoglycemia increases the risk of brain damage from less degrees of hypotension. There are many reports attempting to address the long-term outcomes associated with a glucose concentration of 45 mg/dL versus 47 mg/dL versus 50 mg/dL and all are flawed because none of them look at all three fuels. Thus one cannot draw any firm conclusions from papers currently published about what level of glucose causes brain damage. However, there is evidence to suggest that severe hypoglycemia is associated with a bad outcome with studies that look at patients with a combination of brain imaging abnormalities and poor neurologic outcome finding that 95% of patients had glucose less than 30 mg/dL.
The same may be true for the development of neuroglycopenic symptoms, which typically occur before brain damage would occur. In some disorders, such as defects in ketogenesis, signs and symptoms may begin to appear during fasting at plasma glucose levels of 3.3 mmol/L (60 mg/dL). On the other hand, some patients (those with glucose 6-phosphatase deficiency causing GSD1) may have few symptoms of neuroglycopenia at plasma glucose levels as low as 1.1 to 1.7 mmol/L (20–30 mg/dL) because their high plasma levels of lactate provide an alternative substrate for the brain.
Conditions, such as hyperinsulinism have a very high rate (30%–50%) of brain damage, as they are deficient in glucose and β-hydroxybutyrate and lactate, whereas neonates with GSD1 are often not even diagnosed until they are toddlers and rarely have significant brain damage.
In conclusion, there is no one cutoff for what level of hypoglycemia causes brain damage; rather, it is a complex interplay of fuel availability, oxygen levels, and blood flow that all combine to maintaining the integrity of the brain.
Clinical symptoms and signs associated with hypoglycemia
The clinical features of hypoglycemia in infants may be associated with both neurogenic and neuroglycopenic components ( Box 7.3 ). Symptoms are often subtle and nonspecific, therefore a high index of clinical suspicion must be maintained. Any alteration in clinical status in a newborn that suggests a change in neurologic behavior, fall in temperature, change in feeding pattern, or presence of tremors must be considered a possible initial presentation of a hypoglycemic episode. A seizure or apnea must always be considered a possible manifestation of hypoglycemia and plasma glucose should be measured immediately.
Diagnostic approach
Determining the specific underlying cause of hypoglycemia is critical for the establishment of specific treatment and thus prevention of further episodes of hypoglycemia. The diagnosis should be based on the combination of data obtained from the history, physical examination, laboratory findings, and, especially, the hormonal and fuel responses at the time of hypoglycemia.
Important elements from the pregnancy and perinatal history, in addition to the family history, are critical to determine the existence of risk factors that could point to specific diagnoses, for example perinatal stress-induced hyperinsulinism in a neonate with IUGR. Information regarding the duration of fasting that provoked hypoglycemia is also helpful, for example, onset within a few hours of a meal would be consistent with hyperinsulinism or GSD I, whereas onset after 10 to 12 hours would be consistent with a defect in fatty acid oxidation. Pituitary deficiency with GH or adrenocorticotropic hormone (ACTH)-cortisol deficiency might be suspected by the presence of midline facial malformations, microphthalmia, or microphallus (follicle-stimulating hormone [FSH]/luteinizing hormone [LH] deficiency in utero). Growth failure is also a prominent feature of GSD I or debrancher deficiency glycogen storage disease after the first 3 months of life. Both of these disorders are associated with massive hepatomegaly. Abnormal results of liver function tests (transaminases) and hyperammonemia, with or without elevated creatine kinase level, would suggest a possible fatty acid oxidation disorder (see Table 7.2 ).
A logical diagnostic approach is to analyze a hypoglycemic event as a maladaptation to fasting, and thus the most important information required for diagnosis, comes from tests on the blood and urine specimens obtained at the time of hypoglycemia (also known as the critical samples ). Fig. 7.4 outlines an algorithm for diagnosis of different forms of hypoglycemia based on readily available laboratory tests on the critical samples. The first discriminant is a measure of acidemia at the time of hypoglycemia using the serum bicarbonate. If the acidemia is caused by elevations of the ketoacids (β-hydroxybutyrate and acetoacetate), possibilities include a normal child fasted for too long, a defect in glycogenolysis (glycogen storage disease type 3), or counterregulatory hormone deficiency (hypopituitarism). If the acidemia is caused by an elevation of lactic acid, a block of gluconeogenesis should be suspected (GSD I or fructose 1,6-diphosphatase deficiency or ethanol ingestion). If there is no acidemia (i.e., absence of the normal elevation of ketones) but free fatty acid levels are high, a defect in fatty acid oxidation and ketogenesis should be suspected (e.g., medium-chain acyl-CoA dehydrogenase deficiency [MCAD] deficiency). If ketones are not appropriately increased but the free fatty acid concentrations are also suppressed, hyperinsulinism should be suspected. In the neonatal period, the features of hyperinsulinism can be mimicked by congenital pituitary hormonal deficiency.
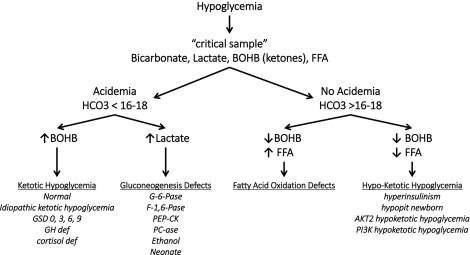
Further tests can then be planned using the initial critical specimens to confirm the suspected diagnosis. These may include physiologic tests (such as the glucagon stimulation test at a time of hypoglycemia to confirm hyperinsulinism) or specialized laboratory tests (such as a plasma acylcarnitine profile) to identify a defect in mitochondrial β-oxidation.
If a critical sample obtained during a spontaneous episode of hypoglycemia is not available, a formal fasting test may be necessary to diagnose the cause of hypoglycemia. The goal of this test is to reproduce the setting in which hypoglycemia occurs to identify the underlying cause. The fasting test should be considered a method of testing a hypothesis that has already been developed, based on available clinical and laboratory data about the cause of the hypoglycemia. Thus the test can be modified with additions or deletions to the basic protocol. This is important, because challenging an infant with fasting is not without risk—particularly if a genetic defect in fatty acid oxidation or adrenal insufficiency is present. Therefore fasting or other diagnostic challenges should be done only in the hospital under carefully controlled settings with an experienced physician and nursing staff readily available. Infants younger than 1 year are usually fasted for up to 24 hours, whereas in older children the maximum fast may be up to 36 hours. The fast is terminated when the plasma glucose falls below 2.8 mmol/L (50 mg/dL), but it may be ended sooner if plasma β-hydroxybutyrate rises to greater than 2.5 mmol/L or if there are any adverse signs or symptoms. Periodic blood samples are obtained for analysis of major fuels and hormones and for appropriate ancillary tests (e.g., serum total carnitine, acylcarnitine profile, liver transaminases, creatine phosphokinase, or urinary organic acids). If hyperinsulinism is suspected, the fasting test may be ended with glucagon (1 mg given IV) to evaluate the glycemic response.
Special note should be made that the most frequent cause of hypoglycemia in neonates, infants, and children (hyperinsulinism) often cannot be diagnosed based solely on the plasma insulin concentration. With very sensitive assays, serum insulin concentrations will be less than 1 to 2 μU/mL at times of hypoglycemia (i.e., below the sensitivity of most insulin assays). However, serum C-peptide concentrations may be a useful marker with values of 0.5 ng/mL or higher strongly suggestive of hyperinsulinism, as shown in Table 7.3 . Therefore the diagnosis must often be made based on evidence of inappropriate insulin effects: hypoketonemia, hypo-free fatty acidemia, and a positive glycemic response to glucagon (see Table 7.3 ).
| Parameter | Sensitivity (%) | Specificity (%) |
|---|---|---|
| Insulin > 2 μU/mL a | 82.2 | 100 |
| C-Peptide ≥ 0.5 ng/mL | 88.5 | 100 |
| β-hydroxybutyrate < 1.8 mmol/L | 100 | 100 |
| Free Fatty Acids < 1.7 mmol/L | 87 | 100 |
| IGFBP-1 ≤ 110 ng/mL | 85 | 96.6 |
| Glycemic response to glucagon > 30 mg/dL | 89 | 100 |
Classification of causes of persistent hypoglycemia in the neonate and infant ( Box 7.4 )
Hyperinsulinism and Similar Disorders
Suppression of insulin secretion is critical for maintaining euglycemia during fasting through the activation of hepatic glycogenolysis, gluconeogenesis, and fatty acid oxidation. Indeed, failure to suppress insulin secretion is the most common cause of persistent hypoglycemia in neonates and children. The clinical and biochemical hallmarks of increased insulin secretion/actions in the neonate are (1) increased birth weight because of the growth-promoting effects of insulin in utero; (2) increased glucose utilization; and (3) evidence that glycogenolysis, lipolysis, and ketogenesis are suppressed, as inferred by the finding of a glycemic response to glucagon (an increase of > 30 mg/dL), and the suppression of plasma free fatty acids and β-hydroxybutyrate concentrations during hypoglycemia (see Table 7.3 ).
- A.
Disorders of Insulin Excess or Actions
- 1.
Hyperinsulinism
- a.
Perinatal Stress-Induced Hyperinsulinism
- b.
Monogenic Hyperinsulinism
K ATP Hyperinsulinism
Focal
Diffuse
GDH Hyperinsulinism
SCHAD Hyperinsulinism
GCK Hyperinsulinism
HNF4A and HNF1A Hyperinsulinism
UCP2 Hyperinsulinism
FOXA2
- c.
Syndromic Hyperinsulinism
Beckwith-Wiedemann Syndrome
Kabuki Syndrome
Turner Syndrome
- a.
- 2.
Postprandial Hyperinsulinemic hypoglycemia after Fundoplication
- 3.
Hypoketotic Hypoglycemia Caused by Activating Mutations in Insulin Signaling Pathway
AKT2-hypoketotic hypoglycemia
PI3K-hypoketotic hypoglycemia
- 1.
- B.
Defects of Counterregulatory Response
- 1.
Hypopituitarism
- 2.
Congenital ACTH Deficiency (TBX19)
- 1.
- C.
Defects in Glycogenolysis and Gluconeogenesis
GSD Type 1
- D.
Defects in Fatty Acid Oxidation
MCAD
- E.
Defects of Glucose Transporters
GLUT1 Deficiency
GLUT2 Deficiency
The lack of alternative fuels (ketones) during hypoglycemia heightens the risk of brain damage in infants with these conditions, thus, it is essential that the diagnosis and establishment of treatment are expedited. The differential diagnosis and mechanisms of disease are broad, but they can be grouped in two categories: (1) hyperinsulinism caused by dysregulated insulin secretion, and (2) mimickers of hyperinsulinism, in which insulin secretion by the pancreatic beta cells is appropriately suppressed during hypoglycemia, but the effects of insulin on insulin-responsive tissues is inappropriately turned on.
Hyperinsulinism
Hyperinsulinism is the most common cause of persistent hypoglycemia in neonates, infants, and children. Hyperinsulinism is caused by dysregulated insulin secretion from the pancreatic β-cells and can be the result of perinatal factors (perinatal stress-induced hyperinsulinism); monogenic, because of mutations in genes important for the regulation of insulin secretion; or syndromic (as in BWS).
Transient Hyperinsulinism Resulting from Maternal Factors
Transient hyperinsulinism is a well-recognized complication in neonates after a pregnancy complicated by gestational diabetes. At birth, infants born to these mothers may be large and plethoric—and their body stores of glycogen, protein, and fat are replete. The classic clinical description of the effect of hyperinsulinism relates to the infant of the diabetic mother:
These infants are remarkable not only because like fetal versions of Shadrack, Meshack, and Abednego, they emerge at least alive from within the fiery metabolic furnace of diabetes mellitus, but because they resemble one another so closely they might well be related. They are plump, sleek, liberally coated with vernix caseosa, full-faced, and plethoric. The umbilical cord and placenta share in the gigantism. During their first 24 or more extrauterine hours, they lie on their backs, bloated and flushed, their legs flexed and abducted, their lightly closed hands on either side of the head, the abdomen prominent and their respiration sighing. They convey a distinct impression of having had such a surfeit of both food and fluid pressed upon them by an insistent hostess that they desire only peace so that they may recover from their excesses. On the second day, their resentment of the slightest noise improves the analogy when their trembling anxiety seems to speak of intrauterine indiscretions of which we know nothing.
The hypoglycemia in infants of diabetic mothers is caused by hyperinsulinemia triggered by maternal hyperglycemia. Infants born to diabetic mothers also have a subnormal surge in plasma glucagon immediately after birth, subnormal glucagon secretion in response to stimuli, and (initially) excessive sympathetic activity that may lead to adrenomedullary exhaustion because urinary excretion of epinephrine is diminished. Thus despite their abundance of tissue stores of available substrate, the normal plasma hormonal pattern of low insulin, high glucagon, and catecholamines is reversed. Their endogenous glucose production is inhibited and glucose utilization is increased compared with that in normal infants, thus predisposing them to hypoglycemia.
Mothers whose diabetes has been well controlled during pregnancy in general, have near-normal-sized infants who are less likely to develop neonatal hypoglycemia and other complications formerly considered typical of maternal diabetes. Nevertheless, treatment of infants born to mothers with diabetes commonly requires provision of IV glucose for a few days, until the hyperinsulinemia abates.
During labor and delivery, maternal hyperglycemia should be avoided because it may result in fetal hyperglycemia—which predisposes to rebound hypoglycemia when the glucose supply is interrupted at birth. Other maternal factors that can result in transient neonatal hyperinsulinism include oral hypoglycemics (such as sulfonylureas) or other medications (terbutaline or propranolol).
By definition, transient hyperinsulinism, as a cause of neonatal hypoglycemia in an infant of a diabetic mother, should abate in 1 or 2 days. If the condition persists, prolonged or congenital hyperinsulinism must be considered.
Perinatal Stress-Induced Hyperinsulinism
The process of beta cell maturation after birth may be impacted by perinatal factors resulting in perinatal stress-induced hyperinsulinism, a distinct form of hyperinsulinism that spontaneously resolves within the first few weeks of life. Perinatal factors associated with perinatal stress-induced hyperinsulinism include birth asphyxia, maternal preeclampsia, prematurity, intrauterine growth retardation, and other peripartum stress. Up to 50% of neonates in these at-risk categories may be affected. The estimated incidence of perinatal stress-induced hyperinsulinism is 1:6,000 to 12,000 live births.
The clinical presentation of perinatal stress-induced hyperinsulinism is characterized by high glucose utilization, and the response to fasting hypoglycemia shows inappropriately detectable plasma insulin, low plasma β-hydroxybutyrate and free fatty acid concentrations, and inappropriate glycemic response to glucagon at the time of hypoglycemia. Unlike the transient hyperinsulinism seen in the infant of the diabetic mother, perinatal stress-induced hyperinsulinism may persist for several weeks. In a series of neonates with perinatal stress-induced hyperinsulinism, the median age of resolution was 6 months. The mechanism responsible for the dysregulated insulin secretion is not known. Acute insulin responses show that, in general, the patterns of insulin response to secretagogues (calcium, tolbutamide, glucose, and leucine) in infants with perinatal stress-induced hyperinsulinism resembled those of normal controls.
Infants with perinatal stress-induced hyperinsulinism usually respond very well to medical therapy with diazoxide. Previously, it was common practice to use pharmacologic doses of glucocorticoids to treat such neonates with persistent hypoglycemia. However, the use of glucocorticoids as nonspecific therapy for neonatal hypoglycemia is not recommended because they are not only ineffective, but also can suppress the hypothalamic-pituitary-adrenal axis.
Monogenic Hyperinsulinism
Congenital hyperinsulinism, also known as persistent hyperinsulinemic hypoglycemia of infancy , represents a group of clinically and genetically heterogeneous disorders characterized by dysregulated insulin secretion and resulting in severe and persistent hypoglycemia. First described in 1954 by MacQuarrie as “idiopathic hypoglycemia of infancy,” congenital hyperinsulinism is the most common cause of persistent hypoglycemia in neonates, infants, and children. Worldwide, the incidence of congenital hyperinsulinism is estimated at 1 in 50,000 live births, with higher incidence of up to 1 in 2500 in areas of high consanguinity.
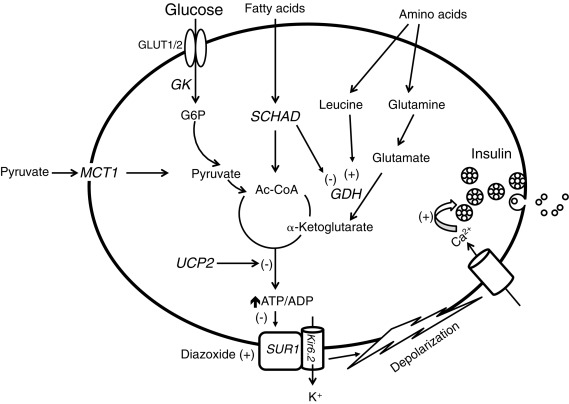
To understand the pathophysiology of congenital hyperinsulinism, knowledge of the major pathways regulating insulin secretion by pancreatic β-cells is critical (outlined in Fig. 7.5 ). Glucose-stimulated insulin secretion involves glucose entry to the beta cell by facilitated diffusion through glucose transporters (mainly GLUT1 in human beta cells) and phosphorylation by glucokinase (GK), leading to glucose oxidation and a rise in the ATP/adenosine diphosphate (ADP) ratio that results in closure of the plasma membrane ATP-dependent potassium (K ATP ) channel. The β-cell K ATP channel is a heterooctameric complex consisting of two subunits: a K + -selective pore-forming subunit (Kir6.2), and a regulatory subunit (sulfonylurea receptor 1 [SUR-1]). Four Kir6.2 subunits form the central pore, coupled to four SUR-1 subunits. The K ATP channel is inhibited (closed) by sulfonylurea drugs (used therapeutically to stimulate insulin secretion in type 2 diabetes), and activated (opened) by diazoxide (the first-line medical treatment for congenital hyperinsulinism). In the unstimulated state, the β-cell ATP-sensitive potassium channels are open—keeping a resting membrane potential of approximately –65 mV. Following the uptake and metabolism of glucose, increase in the intracellular ATP/ADP ratio results in closure of ATP-sensitive potassium channels, depolarization of the cell membrane, opening of voltage-dependent Ca 2 + channels, Ca 2 + influx, rise in cytosolic free Ca 2 + concentration, and activation of the exocytotic machinery. Stimulation of insulin secretion by amino acids occurs through allosteric activation of glutamate dehydrogenase (GDH) by leucine, which results in increased oxidation of glutamate—leading to a rise in the ATP/ADP ratio, inhibition of K ATP -channel activity, and membrane depolarization.