- Iatrogenic hypoglycemia is the limiting factor in the glycemic management of diabetes. It occurs during treatment with an insulin secretagogue or with insulin.
- The key physiologic defenses against falling plasma glucose concentrations are decrements in insulin and increments in glucagon and epinephrine. The behavioral defense is carbohydrate ingestion prompted by symptoms that are largely the result of sympathetic neural activation.
- Hypoglycemia in diabetes is the result of therapeutic hyperinsulinemia. As glucose levels fall, decrements in insulin and increments in glucagon are lost, because of β-cell failure, in type 1 diabetes mellitus (T1DM) and advanced type 2 diabetes mellitus (T2DM). In that setting, attenuated increments in epinephrine cause the syndrome of defective glucose counter-regulation. Attenuated increments in sympathetic neural activity largely bring about the syndrome of hypoglycemia unawareness.
- The concept of hypoglycemia-associated autonomic failure (HAAF) in diabetes posits that recent antecedent hypoglycemia, as well as prior exercise or sleep, causes both defective glucose counter-regulation and hypoglycemia unawareness by attenuating the sympathoadrenal response to subsequent hypoglycemia.
- Hypoglycemia unawareness and, in part, the reduced epinephrine component of defective glucose counter-regulation are reversible in 2–3 weeks with scrupulous avoidance of hypoglycemia in most affected patients.
- The risk factors for hypoglycemia, based on this pathophysiology, include relative as well as absolute therapeutic insulin excess, and factors indicative of HAAF which include absolute endogenous insulin deficiency, a history of severe hypoglycemia, hypoglycemia unawareness, or both as well as prior exercise or sleep, and aggressive glycemic therapy per se.
- This pathophysiology explains why the incidence of iatrogenic hypoglycemia increases over time in T2DM, approaching that in T1DM. Most episodes of hypoglycemia occur in people with T2DM.
- Minimizing the risk of iatrogenic hypoglycemia requires acknowledging the problem, applying the principles of aggressive glycemic therapy and considering the risk factors for relative as well as absolute therapeutic insulin excess and for HAAF.
- Maintenance of the greatest degree of glycemic control that can be accomplished safely in a given patient at a given stage of that individual’s diabetes is in the patient’s best interest. Concerns about hypoglycemia should not be an excuse for poor glycemic control.
Overview of the clinical problem
Iatrogenic hypoglycemia is the most important limiting factor in the glycemic management of diabetes [1,2]. It causes recurrent morbidity in most people with type 1 diabetes mellitus (T1DM) and many with advanced type 2 diabetes mellitus (T2DM), and is sometimes fatal. It compromises physiologic and behavioral defenses against subsequent falling plasma glucose concentrations and thus causes a vicious cycle of recurrent hypoglycemia. It precludes maintenance of euglycemia over a lifetime of diabetes and thus full realization of the vascular benefits of glycemic control.
Hypoglycemia in diabetes is fundamentally iatrogenic, the result of pharmacokinetically imperfect treatments with an insulin secretagogue (e.g. a sulfonylurea or a glinide) or with exogenous insulin that results in episodes of hyperinsulinemia. Nonetheless, hypoglycemia is typically the result of the interplay of relative or absolute therapeutic insulin excess and comprised physiologic and behavioral defenses against falling plasma glucose concentrations [1,2].
The problem of hypoglycemia in diabetes has been summarized [1] and reviewed in detail [2]. My premise is that understanding of the pathophysiology of glucose counter-regulation, the mechanisms that normally prevent or rapidly correct hypoglycemia, leads to insight into the frequency of, risk factors for and prevention of iatrogenic hypoglycemia. Therefore, the physiology of glucose counter-regulation and its pathophysiology in diabetes are addressed first, followed by a summary of the magnitude of the clinical problem. With that background, a clinical approach to the prevention of iatrogenic hypoglycemia, and if necessary its treatment, is discussed.
Physiology of glucose counter-regulation
Hypoglycemia and the brain
Glucose is an obligate oxidative fuel for the brain under physiologic conditions [1–4]. The brain accounts for more than 50% of whole body glucose utilization. Because it cannot synthesize glucose, utilize physiologic concentrations of circulating non-glucose fuels effectively or store more than a few minutes supply as glycogen [1,2], the brain requires a virtually continuous supply of glucose from the circulation. Because facilitated blood–brain glucose transport is a direct function of the arterial plasma glucose concentration, that requires maintenance of the physiologic plasma glucose concentration. Hypoglycemia causes functional brain failure, which is typically reversed after the plasma glucose concentration is raised [4]. Rarely, profound prolonged hypoglycemic causes brain death [4].
Responses to hypoglycemia
Falling plasma glucose concentrations cause a sequence of responses in individuals without diabetes (Table 33.1) [1,5–8]. The first physiologic response, which occurs as plasma glucose concentrations decline within the postabsorptive plasma glucose concentration range, is a decrease in insulin secretion. The secretion of glucose counterregulatory (plasma glucose raising) hormones, including glucagon and epinephrine, increases as plasma glucose concentrations fall just below the physiologic range. Lower plasma glucose levels cause a more intense sympathoadrenal – sympathetic neural as well as adrenomedullary – response and symptoms. Even lower plasma glucose concentrations cause functional brain failure [4].
Table 33.1 Physiologic responses to decreasing plasma glucose concentrations.
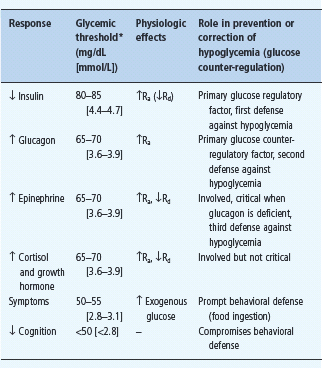
Ra, rate of glucose appearance, glucose production by the liver and kidneys;
Rd rate of glucose disappearance, glucose utilization by insulin-sensitive tissues such as skeletal muscle (no direct effect on central nervous system glucose utilization).
* Arterialized venous, not venous, plasma glucose concentrations. Reproduced from Cryer PE. Glucose homeostasis and hypoglycemia. In: Kronenberg HM, Melmed S, Polonsky KS, Larsen PR, eds. Williams Textbook of Endocrinology, 11th edn. Philadelphia: Saunders, 2008: 1503–1533, with permission.
Clinical manifestations of hypoglycemia
The symptoms and signs of hypoglycemia are not specific [8,9]. Thus, clinical hypoglycemia is most convincingly documented by Whipple’s triad: symptoms, signs, or both, consistent with hypoglycemia; a low measured plasma glucose concentration; and resolution of those symptoms and signs after the plasma glucose concentration is raised.
Neuroglycopenic symptoms, which are the result of brain glucose deprivation per se, include cognitive impairments, behavioral changes and psychomotor abnormalities and, at lower plasma glucose concentrations, seizure and coma [1,2,4,8,9]. Neurogenic (or autonomic) symptoms, which are largely the result of the perception of physiologic changes caused by the sympathoadrenal – particularly the sympathetic neural [9] – discharge triggered by hypoglycemia, include both adrenergic (e.g. palpitations, tremulousness and anxiety/arousal) and cholinergic (e.g. sweating, hunger and paresthesias) symptoms [8]. Central mechanisms may also be involved in some of the latter symptoms such as hunger [10]. Awareness of hypoglycemia is largely the result of the perception of neurogenic symptoms [8].
Signs of hypoglycemia include pallor and diaphoresis, the result of adrenergic cutaneous vasoconstriction and cholinergic activation of sweat glands, respectively [1,2]. Neuroglycopenic manifestations are often observable.
Maintenance of systemic glucose balance
Because obligatory glucose utilization, largely by the brain, is fixed and exogenous glucose delivery from ingested carbohydrates is intermittent, systemic glucose balance is maintained, and hypoglycemia (as well as hyperglycemia) is prevented, by dynamic regulation of endogenous glucose production from the liver (and the kidneys) and of glucose utilization by non-neural tissues such as muscle [1,11]. Although an array of hormones, neurotransmitters and metabolic substrates are involved, endogenous glucose production and glucose utilization by non-neural tissues are regulated primarily by insulin (Figure 33.1; Table 33.1) [11].
Figure 33.1 Physiologic and behavioral defenses against hypoglycemia in humans. ACh, acetylcholine; α-cell, pancreatic islet β-cell; β-cell, pancreatic islet β-cell; NE, norepinephrine; PNS, parasympathetic nervous system; SNS, sympathetic nervous system; T1DM, type 1 diabetes mellitus. Reproduced from Cryer PE. Mechanisms of sympathoadrenal failure and hypoglycemia in diabetes. J Clin Invest 2006; 116:1470–1473, with permission from the American Society for Clinical Investigation.
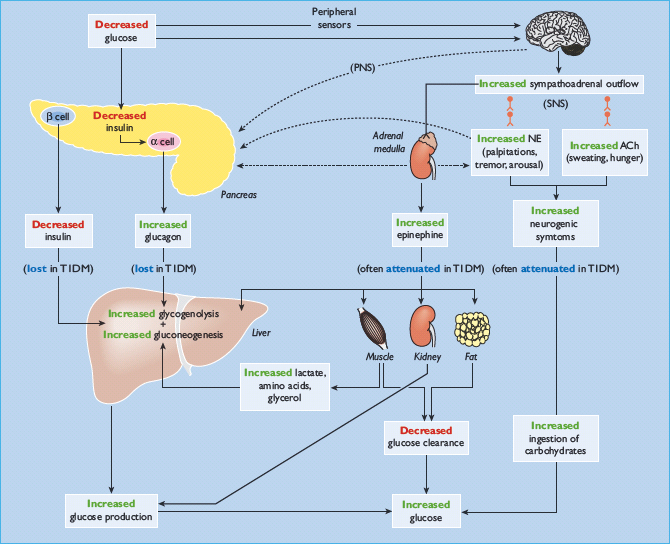
The first physiologic defense against hypoglycemia is a decrease in pancreatic islet β-cell insulin secretion. That occurs as plasma glucose concentrations decline within the physiologic range (Table 33.1) and favors increased hepatic (and renal) glucose production with virtual cessation of glucose utilization by insulin-sensitive tissues such as muscle (Figure 33.1). The second physiologic defense is an increase in pancreatic islet α-cell glucagon secretion. That occurs as plasma glucose concentrations fall just below the physiologic range (Table 33.1) and stimulates hepatic glucose production (largely by stimulating glycogenolysis) (Figure 33.1). Increased glucagon secretion is signaled primarily by a decrease in intra-islet insulin, perhaps among other β-cell secretory products, in the setting of low glucose concentrations [1,2,12] and secondarily by increased autonomic nervous system – sympathetic, parasympathetic and adrenomedullary – inputs [1,2,13]. The third physiologic defense, which becomes critical when glucagon is deficient, is an increase in adrenomedul-lary epinephrine secretion [1,2,11]. That, too, occurs as plasma glucose concentrations fall just below the physiologic range (Table 33.1). It raises plasma glucose concentrations largely by β2-adrenergic stimulation of hepatic (and renal) glucose production (Figure 33.1), but the glucose-raising actions of epinephrine also involve limitation of glucose clearance by insulin sensitive tissues, mobilization of gluconeogenic substrates such as lactate and amino acids from muscle and glycerol from fat and α2-adrenergic limitation of insulin secretion [1,2,14]. Sympathoadrenal activity, including epinephrine secretion, is regulated in the brain [1,2,11]. Circulating epinephrine is almost exclusively derived from the adrenal medulle [9]; while circulating norepinephrine is largely derived from sympathetic nerves under resting and many stimulated (e.g. exercise) conditions, the plasma norepinephrine response to hypoglycemia is also largely adrenomedullary in origin [9].
If these physiologic defenses fail to abort an episode of developing hypoglycemia, lower plasma glucose concentrations cause a more intense sympathoadrenal response that causes neurogenic symptoms (Figure 33.1) [1,2,11]. Those symptoms cause awareness of hypoglycemia that prompts the behavioral defense of ingestion of carbohydrates [1,2,11].
All of these defenses against hypoglycemia, not just insulin secretion, are typically compromised in people with T1DM and those with advanced (i.e. absolutely endogenous insulin deficient) T2DM (Figure 33.1) [1,2].
Pathophysiology of glucose counter-regulation in diabetes
Insulin excess
While substantial insulin excess alone can cause hypoglycemia, the integrity of the defenses against falling plasma glucose concentrations determines whether a typical episode of therapeutic hyperinsulinemia results in clinical hypoglycemia [1,2]. Thus, iatrogenic hypoglycemia is typically the result of the interplay of relative or absolute therapeutic hyperinsulinemia and compromised physiologic and behavioral defenses against falling plasma glucose concentrations (Figure 33.1; Table 33.2) [1,2,15,16].
Table 33.2 Responses to falling plasma glucose concentrations in humans.
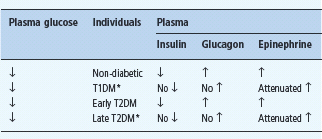
* These alterations account for the appearance of defective glucose counter – regulation and hypoglycemia unawareness in T1DM and late T2DM.
Defective glucose counter-regulation and hypoglycemia unawareness
In fully developed (i.e. C peptide negative) T1DM circulating insulin concentrations cannot decrease as plasma glucose concentrations fall in response to therapeutic (exogenous) hyperinsulinemia (Table 33.2) [1,2,15]. That is the result of β-cell failure which also causes loss of the increase in circulating glucagon concentrations (Figure 33.2; Table 33.2) [1,2,12,15]. The latter conclusion is based on the finding that indirect reciprocal β-cell-mediated signaling normally predominates over direct α-cell signaling in the regulation of glucagon secretion in humans [1,2,12]. Thus, the absence of a decrease in intra-islet insulin, perhaps among other β-cell secretory products, in concert with low glucose levels, plausibly explains loss of the glucagon response [1,2,12]. Therefore, both the first and second physiologic defenses against hypoglycemia are lost. Furthermore, the increase in circulating epinephrine, the third physiologic defense, is typically attenuated (Figure 33.2; Table 33.2) [1,15]. In the setting of absent insulin and glucagon responses, the attenuated epinephrine response causes the clinical syndrome of defective glucose counter-regulation (Table 33.2) [1,2,15,17,18] which is associated with a 25-fold [17] or greater [18] increased risk of severe iatrogenic hypoglycemia in T1DM. In addition, the attenuated epinephrine response is a marker for an attenuated sympathoad-renal, including sympathetic neural, response (Table 33.2) which is largely responsible for reduced neurogenic symptom responses and therefore the clinical syndrome of hypoglycemia unaware-ness [1,2,9] which is associated with a sixfold increased risk of severe iatrogenic hypoglycemia in T1DM [19]. Attenuated sympathoadrenal responses to falling plasma glucose concentrations can be caused by recent antecedent hypoglycemia (Figures 33.3 and 33.4) [1,2,15,16,20], prior exercise [1,2,21–23] or sleep [1,2,24–26].
Figure 33.2 Mean (± SE) plasma glucose, insulin, epinephrine and glucagon concentrations during hyperinsulinemic stepped hypoglycemic glucose clamps in individuals without diabetes (red squares and columns), people with type 1 diabetes (T1DM, insulin – dependent diabetes mellitus) with classic diabetic autonomic neuropathy (CDAN) (yellow triangles and columns) and people with type 1 diabetes without CDAN (blue circles and columns). Reproduced from Dagogo-Jack et al. [15] with permission from the American Society for Clinical Investigation.
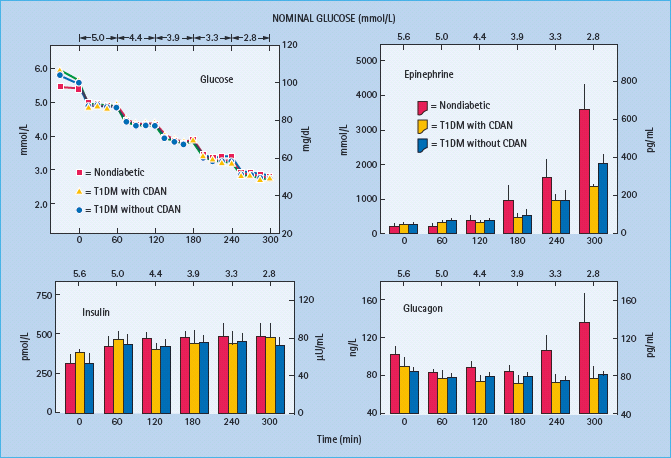
Figure 33.3 Mean (± SE) plasma glucose, insulin, epinephrine and glucagon concentrations during hyperinsulinemic stepped hypoglycemic glucose clamps in patients with type 1 diabetes without classic diabetic autonomic neuropathy on mornings following afternoon hyperglycemia (red circles and columns) and on mornings following afternoon hypoglycemia (yellow circles and columns). From Dagogo-Jack et al. [15] with permission from the American Society for Clinical Investigation.
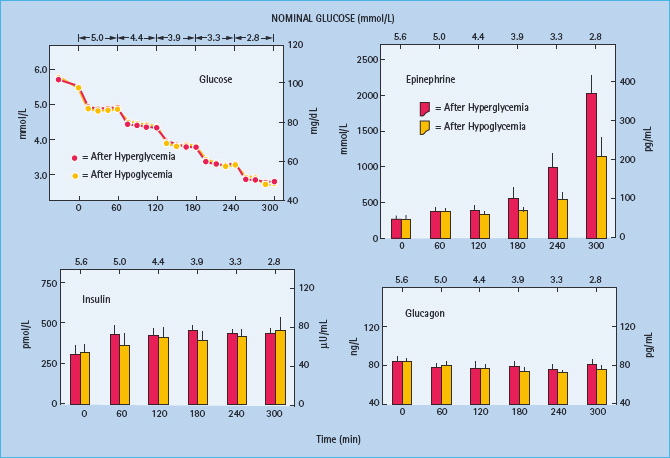
Figure 33.4 Mean (± SE) total, neurogenic and neuroglycopenic symptom scores during hyperinsulinemic stepped hypoglycemic clamps in patients with type 1 diabetes without classic diabetic autonomic neuropathy on mornings following afternoon hyperglycemia (red columns) and on mornings following afternoon hypoglycemia (yellow columns). Reproduced from Dagogo-Jack et al. [15] with permission from the American Society for Clinical Investigation.
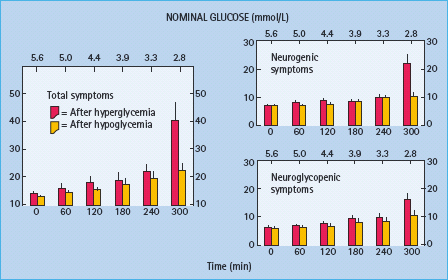
While hypoglycemia unawareness is largely the result of reduced release of the neurotransmitters norepinephrine and ace-tylcholine [1,9], there is evidence of decreased β-adrenergic sensitivity, specifically reduced cardiac chronotropic sensitivity to isoproterenol, in affected patients [27,28]. However, vascular sensitivity to β2-adrenergic agonism was not found to be reduced in patients with unawareness [29], reduced sensitivity to β-adrenergic signaling of neurogenic symptoms remains to be demonstrated in patients with unawareness and it would be necessary to postulate decreased cholinergic sensitivity as well to explain reduced cholinergic symptoms such as sweating.
The pathophysiology of glucose counterregulation is the same in T1DM and advanced (i.e. absolutely endogenous insulin deficient) T2DM, albeit with different time courses (Table 33.2) [1,2,15,16]. The pathogenesis of an episode of iatrogenic hypoglycemia involves therapeutic hyperinsulinemia resulting in falling plasma glucose concentrations and loss of the appropriate decrements in insulin and increments in glucagon. The hypoglycemia, in turn, reduces the sympathoadrenal responses to subsequent hypoglycemia. Because β-cell failure, which causes loss of both the insulin and the glucagon responses [1,2,15,16], occurs rapidly in T1DM but slowly in T2DM, the syndromes of defective glucose counterregulation and hypoglycemia unawareness develop early in T1DM but later in T2DM. This temporal pattern of compromised glycemic defenses explains why iatrogenic hypoglycemia becomes progressively more frequent as patients approach the insulin deficient end of the spectrum of T2DM as discussed later in this chapter.
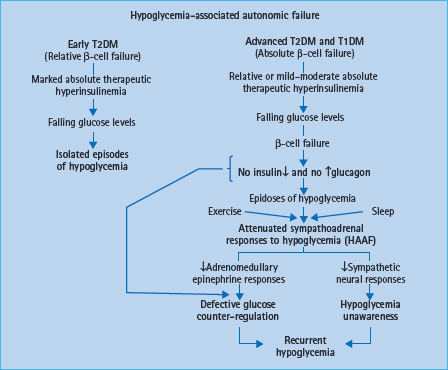
Hypoglycemia-associated autonomic failure
The concept of hypoglycemia-associated autonomic failure (HAAF) in diabetes posits that recent antecedent hypoglycemia (Figures 33.3 and 33.4) [1,2,15,16,20] – as well as prior exercise [1,2,21–23] or sleep [1,2,24–26] – causes both defective glucose counter-regulation (by attenuating the epinephrine responses to subsequent hypoglycemia in the setting of absent decrements in insulin and absent increments in glucagon) and hypoglycemia unawareness (by attenuating the sympathoadrenal, including the sympathetic neural, and resulting neurogenic symptom responses to subsequent hypoglycemia). It therefore leads to a vicious cycle of recurrent iatrogenic hypoglycemia (Figure 33.5). Thus, HAAF is, at least in large part, a dynamic functional disorder that is distinct from classic diabetic autonomic neuropathy [1,2]. Nonetheless, the key feature of HAAF, an attenuated sympathoadrenal response to a given level of hypoglycemia, is more prominent in patients with diabetic autonomic neuropathy (Figure 33.2) [30,31].
Figure 33.5 Hypoglycemia-associated autonomic failure (HAAF) in the pathogenesis of iatrogenic hypoglycemia in diabetes.
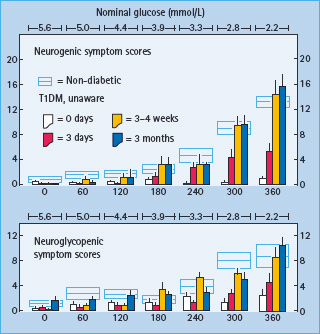
The clinical impact of HAAF is well established in T1DM [1,2,15,32–37]. Recent antecedent hypoglycemia, even asymptomatic nocturnal hypoglycemia, reduces epinephrine, symptomatic and cognitive dysfunction responses to a given level of subsequent hypoglycemia [32]. reduces detection of hypoglycemia in the clinical setting [33] and reduces defense against hyperinsulinemia [15] in T1DM. Perhaps the most compelling support for the concept of HAAF is the finding, initially by three independent research teams [34–37], that as little as 2–3 weeks of scrupulous avoidance of hypoglycemia reverses hypoglycemia unawareness (Figure 33.6) and improves the attenuated epinephrine component of defective glucose counter-regulation in most affected patients.
Figure 33.6 Mean (± SE) neurogenic and neuroglycopenic symptom scores during hyperinsulinemic stepped hypoglycemic clamps in individuals without diabetes (open rectangles) and in patients with T1DM (columns) at baseline (0 days), after 3 days of inpatient strict avoidance of hypoglycemia and after 3±4 weeks and 3 months of outpatient scrupulous avoidance of hypoglycemia. From Dagogo-Jack et al. [37] with permission from the American Diabetes Association.
People with advanced T2DM are also at risk for HAAF [1,2,16]. Glucagon responses to hypoglycemia are lost [16], as they are in T1DM. Furthermore, the glycemic thresholds for sympathoadrenal and symptomatic (among other) responses to hypoglycemia are shifted to lower plasma glucose concentrations by recent antecedent hypoglycemia [16], as they are in T1DM.
The three recognized causes of HAAF each cause attenuated sympathoadrenal and symptomatic (among other) responses to a given level of hypoglycemia [1,2]. Antecedent hypoglycemia-related HAAF [1,2,15,16,20] led to the concept. Exercise-related HAAF [1,2,21–23] is exemplified by late post-exercise hypoglycemia which typically occurs 6–15 hours after strenuous exercise and is often nocturnal [38,39]. Sleep-related HAAF [1,2,24–26] is the result of further attenuation of the sympathoadrenal response to hypoglycemia during sleep. Sleeping patients are therefore much less likely to be awakened by hypoglycemia than individuals without diabetes [25,26]. There may well be additional, as yet unrecognized, functional, and therefore potentially reversible, causes of HAAF [1,2]. In addition, there may be a structural component [1,2].
The mechanisms of HAAF are summarized in Figure 33.7 [1,2]. Loss of the insulin and glucagon responses to falling plasma glucose concentrations caused by therapeutic hyperinsulinemia is the result of β-cell failure in T1DM and advanced T2DM. Because normal insulin and glucagon responses to low glucose concentrations occur in patients with a transplanted (i.e. dener-vated) pancreas [40] and in dogs with a denervated pancreas [41] – as well as from the perfused pancreas and perifused pancreatic islets – innervation is not required.
Figure 33.7 Pancreatic islet, hypothalamic and cerebral network mechanisms of hypoglycemia-associated autonomic failure (HAAF) in diabetes. Reproduced from Cryer [1] with permission from the American Diabetes Association.
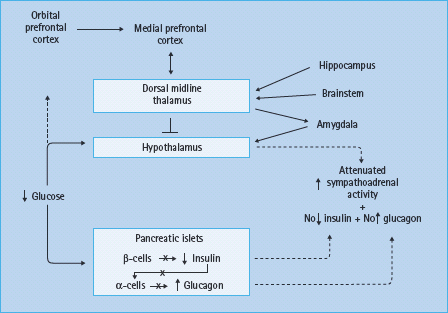
In the setting of absent insulin and glucagon responses to falling plasma glucose concentrations, attenuated sympathoadrenal responses cause both defective glucose counter-regulation and hypoglycemia unawareness, the two components of HAAF [1,2]. The mechanism of the attenuated sympathoadrenal response is not known, but it must be at the level of the brain (or the afferent or efferent components of the sympathoadrenal system) (Figure 33.7). The proposed mechanisms include the systemic mediator, brain fuel transport and brain metabolism hypotheses, all of which have been reviewed [1,2,42,43].
Much of the research into the pathogenesis of HAAF has focused on the hypothalamus, the central integrator of the sympathoadrenal responses to hypoglycemia [43]. While the primary alteration could reside in the hypothalamus, the changes in hypothalamic function could be secondary to those in other brain regions. For example, measurements of regional cerebral blood flow with [15O] water and positron emission tomography (PET) [44] indicate that hypoglycemia activates widespread but interconnected brain regions, including the medial prefrontal cortex, the lateral orbitofrontal cortex, the thalamus, the globus pallidus and the periaqueductal gray. These studies also show that recent antecedent hypoglycemia both reduces the sympathoadrenal and symptomatic responses (a model of HAAF) and causes a greater increase in synaptic activity in the dorsal midline thalamus during subsequent hypoglycemia [45]. Thus, it has been suggested that there may be a cerebral network that results in thalamic inhibition of hypothalamic activity in HAAF (Figure 33.7) [45]. That suggestion is generically consistent with the findings of various patterns of [18F] deoxyglucose uptake in T1DM patients with and without hypoglycemia unawareness [46].
Risk factors for hypoglycemia in diabetes
The risk factors for hypoglycemia in diabetes follow directly from the pathophysiology of glucose counter-regulation and are based on the principle that iatrogenic hypoglycemia is typically the result of the interplay of relative or absolute therapeutic insulin excess and compromised physiologic and behavioral defenses against falling plasma glucose concentrations (i.e. HAAF) in T1DM and advanced T2DM [1,2,47].
Absolute or relative insulin excess
The conventional risk factors for hypoglycemia in diabetes are based on the premise that absolute or relative therapeutic insulin excess is the sole determinant of risk (Table 33.3) [1,2,47]. Absolute therapeutic insulin excess occurs when insulin secretagogue or insulin doses are excessive, ill- timed or of the wrong type, or when insulin clearance is reduced as in renal failure. Relative therapeutic insulin excess occurs under a variety of conditions. It occurs when exogenous glucose delivery is decreased (as following missed or low carbohydrate meals and during the overnight fast), when glucose utilization is increased (as during and shortly after exercise), when endogenous glucose production is decreased (as following alcohol ingestion) and when sensitivity to insulin is increased (as following weight loss or improved glycemic control and in the middle of the night). People with diabetes and their caregivers must consider each of these risk factors when the problem of iatrogenic hypoglycemia is recognized. Overall, however, they explain only a minority of episodes of hypoglycemia [48].
Table 33.3 Risk factors for hypoglycemia in diabetes.
| Relative or absolute insulin excess 1 Insulin or insulin secretagogue doses are excessive, ill-timed or of the wrong type 2 Exogenous glucose delivery is decreased (e.g. following missed meals and during the overnight fast) 3 Glucose utilization is increased (e.g. during and shortly after exercise) 4 Endogenous glucose production is decreased (e.g. following alcohol ingestion) 5 Sensitivity to insulin is increased (e.g. in the middle of the night and following weight loss or improved glycemic control) 6 Insulin clearance is decreased (e.g. with renal failure) |
| Hypoglycemia-associated autonomic failure 1Absolute endogenous insulin deficiency 2A history of severe hypoglycemia, hypoglycemia unawareness, or both as well as recent antecedent hypoglycemia, prior exercise and sleep 3Aggressive glycemic therapy per se (lower HbA1c levels, lower glycemic goals) |
Compromised defenses against hypoglycemia
The risk factors indicative of HAAF (Table 33.3) [1,2,47,49–55] include absolute endogenous insulin deficiency [1,2,47,49–53], a history of severe iatrogenic hypoglycemia, hypoglycemia unawareness, or both as well as recent antecedent hypoglycemia, prior exercise or sleep [1,2,47,50–52,54] and aggressive glycemic therapy per se (i.e. lower HbA1c levels, lower glycemic goals, or both) [1,2,47,50–55]. The degree of endogenous insulin deficiency (i.e. β-cell failure) determines the extent to which insulin levels will not decrease and glucagon levels will not increase as plasma glucose concentrations fall in response to therapeutic hyperinsulinemia. A history of severe hypoglycemia indicates, and that of hypoglycemia unawareness implies, recent antecedent hypoglycemia. The latter causes attenuated sympathoadrenal and symptomatic responses to subsequent hypoglycemia, the key feature of HAAF. In addition, prior exercise and sleep cause that feature of HAAF. Studies of intensive glycemic therapy with a control group treated to a higher HbA1c level consistently report higher rates of hypoglycemia in the group treated to lower HbA1c levels in T1DM [56–58] and T2DM [54,59,60]. That does not mean that one cannot both improve glycemic control and minimize the risk of hypoglycemia [1,2,47] as discussed below.
Magnitude of the clinical problem of hypoglycemia in diabetes
Diabetes is an increasingly common disease. It has been estimated that its prevalence will rise from 285 million people in the year 2009 to 435 million people by the year 2030 [61]. Iatrogenic hypoglycemia affects most of the minority with T1DM and many of the majority with T2DM [1,2]. Indeed, because it precludes maintenance of euglycemia over a lifetime of diabetes, the barrier of hypoglycemia ultimately affects all people with diabetes [1,2].
Frequency of hypoglycemia
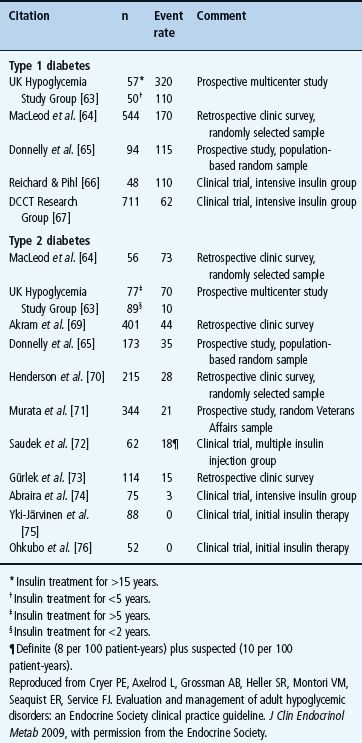
Hypoglycemia is a fact of life for people with T1DM (Table 33.4) [1,2,55,62,63]. The average patient has untold numbers of episodes of asymptomatic hypoglycemia and experiences two episodes of symptomatic hypoglycemia per week – thousands of such episodes over a lifetime of diabetes – and one or more episodes of severe, temporarily disabling hypoglycemia, often with seizure or coma, per year. There is no evidence that this problem has abated since it was highlighted by the report of the Diabetes Control and Complications Trial (DCCT) in 1993 [56]. For example, in 2007 the UK Hypoglycemia Study Group [63] reported an incidence of severe hypoglycemia that was twice that in the DCCT in patients with T1DM for <5 years and an incidence fivefold higher than that in the DCCT in those with T1DM for >15 years (Table 33.4). An incidence comparable to the latter was also found in a large observational study [55].
Table 33.4 Event rates for severe hypoglycemia (that requiring the assistance of another person), expressed as episodes per 100 patient-years, in insulin-treated diabetes [2] .
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


