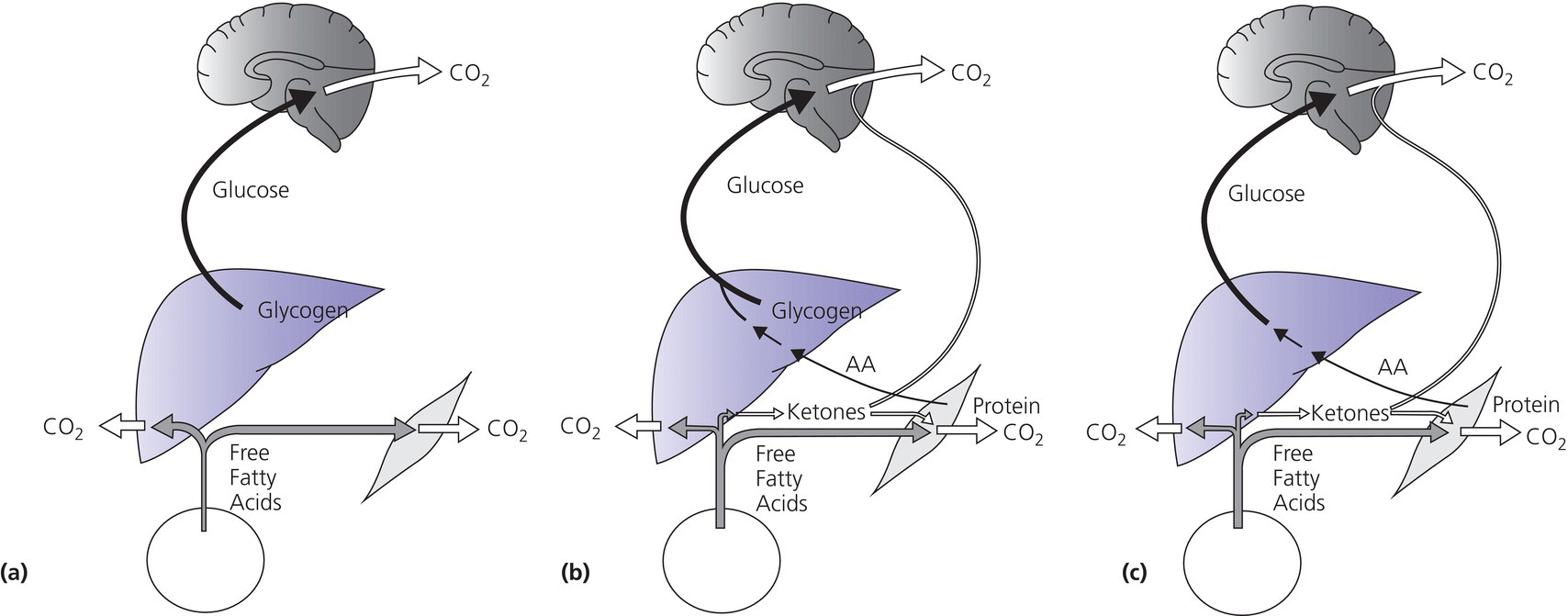
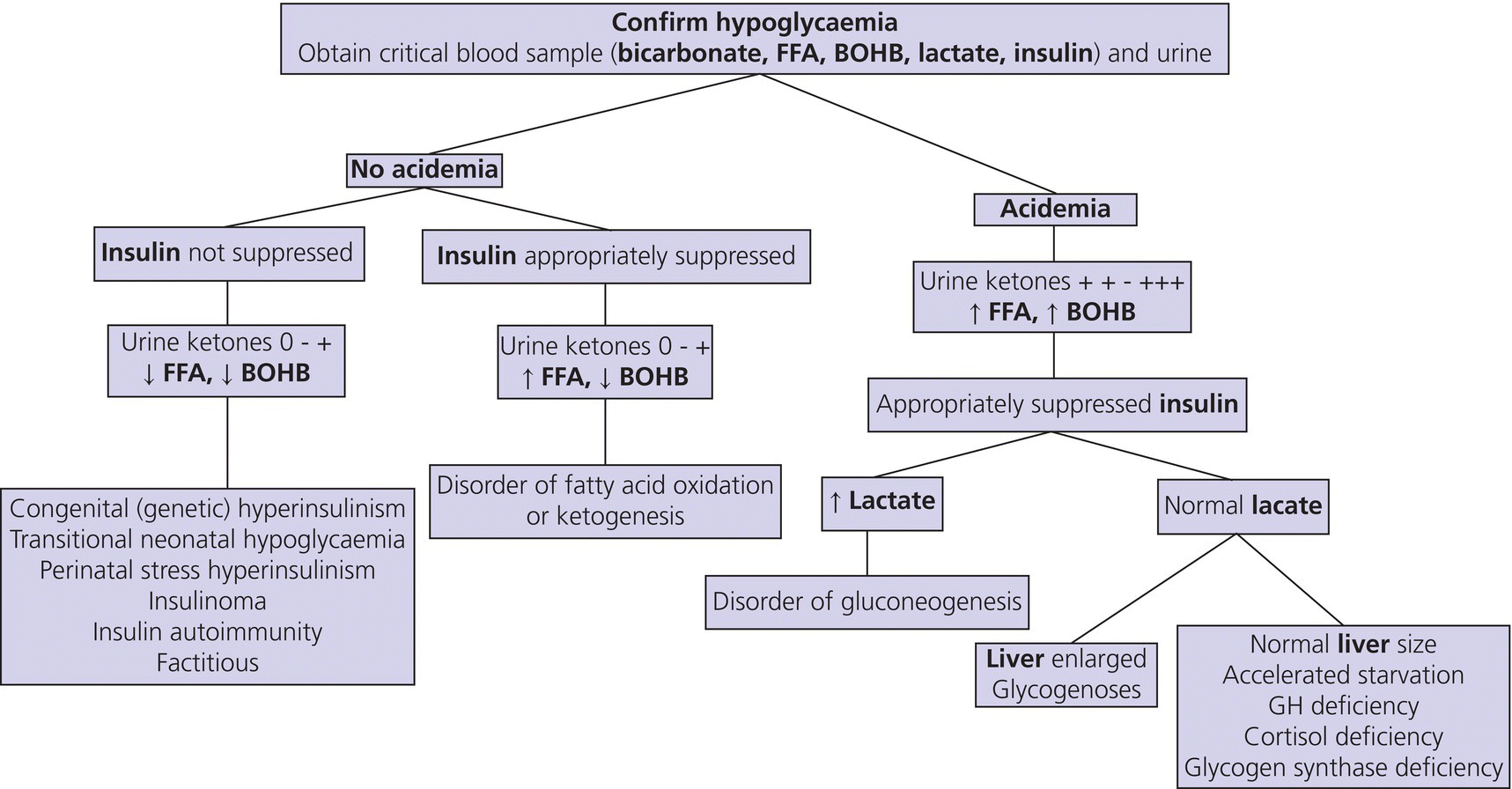
2 Glucose is the body’s main fuel and the brain is the principal consumer of circulating glucose. The brain can neither synthesize glucose nor store more than a few minutes’ supply as glycogen and depends on a continuous supply of glucose. Blood glucose concentrations are normally maintained within a relatively narrow range. Recurrent or persistent hypoglycaemia during the period of rapid brain growth and differentiation in infancy can cause permanent neurological sequelae, psychomotor retardation, and seizures. Prevention of hypoglycaemia and its prompt recognition, accurate diagnosis, and vigorous treatment are essential to prevent its devastating cerebral consequences. Hypoglycaemia most often occurs in the newborn period and is the most common metabolic disorder of the newborn (see Table 2.1 showing neonates at increased risk for hypoglycaemia). The ratio of surface area to body mass of a full‐term newborn baby is approximately three times that of an average adult, necessitating a high rate of energy expenditure to maintain body temperature. Also, the infant brain is large relative to its body mass (and hepatic glycogen stores) and its energy requirement is primarily derived from oxidation of circulating glucose. To meet the high demand for glucose, the rate of glucose production in newborn infants and young children (5–8 mg per kg per minute) is two to three times that of older children and adults (2.3 mg per kg per minute). As a consequence, until feeding is well established, maintenance of glucose homeostasis in the newborn period is more precarious than later in life. During infancy and childhood, hypoglycaemia most often occurs when night‐time feeding is discontinued or when an intercurrent illness interrupts a child’s normal feeding pattern, resulting in a period of relative starvation. Table 2.1 Neonates at increased risk for hypoglycaemia and who require glucose screening. During prolonged fasting, infants and young children cannot sustain the high rate of glucose production. Compared to older children, adolescents, and adults, plasma glucose decreases and plasma ketone concentrations increase more rapidly. In response to feeding, blood glucose concentrations increase, which stimulates insulin and inhibits glucagon secretion, to maintain plasma glucose concentrations below 7–9 mmol/L [126–162 mg/dL]. Insulin increases glucose uptake in skeletal muscle and adipose tissue by increasing GLUT4 expression, promotes hepatic and skeletal muscle glycogen synthesis, and inhibits glycogenolysis and gluconeogenesis. During fasting, plasma glucose concentrations decrease, insulin secretion is suppressed, and increased secretion of the counter‐regulatory hormones glucagon, adrenaline (epinephrine), noradrenaline (norepinephrine), cortisol, and growth hormone (GH) prevent hypoglycaemia. The endocrine and metabolic responses involved in the transition from the fed to the fasted state are shown in Figure 2.1. Figure 2.1 Transition from the fed to the fasting state. (a) Between meals: glucose is from hepatic glycogenolysis; free fatty acids from adipose tissue are an additional source of fuel for muscle. (b) Overnight fast: liver glycogen is depleted and gluconeogenesis becomes the principal source of glucose. Hepatic ketone production increases and rising plasma ketone body concentrations provide an alternative fuel for brain and muscle. (c) Prolonged starvation: fat‐derived fuels are the predominant metabolic substrates. Brain utilization of ketones increases. Glucose is from gluconeogenesis. Source: Pediatric Endocrinology, ed. Fima Lifshitz, 5th edition, 2007. In healthy children, glycogen stores are largely depleted within approximately 12 hours of fasting. Thereafter, gluconeogenesis (from amino acids, glycerol, and lactate) is the sole source of glucose. Lipolysis is activated, increasing the supply of free fatty acids, an alternative energy source for most tissues, which reduces glucose consumption. The brain does not oxidize free fatty acids. However, water‐soluble acetoacetate and ß‐hydroxybutyrate (ketones), produced by hepatic ß‐oxidation of fatty acids, readily cross the blood‐brain barrier and become a major source of energy for the brain during prolonged fasting. During progressively severe hypoglycaemia, a hierarchy of physiological responses occurs, from sympathoadrenal activation to neuroglycopenia. Symptoms and signs are non‐specific and reflect responses of the central nervous system to glucose deprivation (Table 2.2). Neurogenic (autonomic) symptoms result from the central nervous system‐mediated sympathoadrenal discharge triggered by hypoglycaemia. Neuroglycopenic symptoms are caused by brain dysfunction from impaired energy metabolism, i.e. the direct result of brain glucose deprivation. Symptoms are particularly difficult to identify in newborns, who are at greatest risk of developing hypoglycaemia. Neonates with low blood glucose concentrations are frequently asymptomatic and hypoglycaemia is usually detected by routine screening of at‐risk infants or as an incidental laboratory finding. With suspected hypoglycaemia, it is essential to accurately measure a confirmatory plasma or blood glucose concentration and demonstrate that administration of glucose promptly relieves the symptoms and signs (Whipple’s triad, see section ‘Definition’). Table 2.2 Symptoms and signs of hypoglycaemia in newborns. Additional signs of hypoglycaemia in newborns may include apnoea, bradycardia, cyanosis, and hypothermia. In older individuals, typical adrenergic symptoms include palpitations, tremor, and anxiety; cholinergic symptoms include sweating, hunger, and paresthesias. Typical signs include pallor, diaphoresis, tachycardia, and elevated systolic blood pressure. The numerical definition of hypoglycaemia continues to be controversial, most especially in newborns, related to uncertainty regarding the definition of euglycaemia and the glucose threshold resulting in brain injury. In the absence of a consensus, several methods are employed to pragmatically define hypoglycaemia, including statistical, metabolic, neurophysiological, and neurodevelopmental (i.e. evidence that a particular blood glucose concentration is associated with long‐term neurodevelopmental sequelae). Clinical hypoglycaemia is defined as a plasma glucose concentration low enough to cause symptoms and/or signs of impaired brain function. This glucose concentration varies from one patient to another and depends on the availability of alternative fuels and previous blood glucose concentrations. Hypoglycaemia may be difficult to recognize, especially in neonates and young children in whom the symptoms and signs are not specific. Plasma glucose concentrations are also influenced by several factors, including when the sample is obtained relative to the last meal, the source of blood (arterial, capillary, or venous), and whether measurements are performed on whole blood, serum, or plasma (approximately 12–15% higher than whole blood). Furthermore, a single low plasma glucose concentration, especially when measured on a point‐of‐care device (glucose meter), may be a measurement artefact. Confirmation of Whipple’s triad is valuable in older children: (i) symptoms and/or signs consistent with hypoglycaemia; (ii) an accurately measured low plasma glucose concentration; and (iii) relief of symptoms and signs when plasma glucose concentration is restored to normal. Because reliance on Whipple’s triad is not possible in infants and young children, recognition of hypoglycaemia may require confirmation by repeated measurements of plasma glucose concentration and, in certain circumstances, by performing a controlled fasting test. In adults, the glycaemic threshold for a decrease in brain glucose metabolism is normally <54 mg/dL [3.0 mmol/L]. During acute insulin‐induced hypoglycaemia, autonomic symptoms appear at a plasma glucose of approximately 60 mg/dL [3.3 mmol/L] and impaired brain function occurs at approximately 50 mg/dl [2.8 mmol/L]. In infants and children, functional changes in the central nervous system (brainstem and somatosensory evoked potentials) occur when the venous plasma glucose falls below 47 mg/dL [2.6 mmol/L]. These observations suggest that the physiologic threshold is a plasma glucose in the range 50–60 mg/dL [2.8–3.3 mmol/L]. For clinical care of children, a venous plasma glucose of ≥60 mg/dL [3.3 mmol/L] may be regarded as normoglycaemia and levels <50 mg/dL [2.8 mmol/L] as hypoglycaemia. When considering management decisions, the above numerical definitions are useful; however, plasma glucose concentrations should also be evaluated in the context of the availability of other metabolic fuels (lactate and ketones). For example, a plasma glucose concentration <50 mg/dL [2.8 mmol/L] is not necessarily harmful if the blood ketone concentration is markedly elevated because infants and children can utilize ketones for cerebral metabolism. Note, however, that free fatty acid and ketone levels are suppressed by insulin, thus accounting for the increased risk of brain injury in infants with hyperinsulinaemic hypoglycaemia. Hypoglycaemia occurs when the rate of blood glucose utilization exceeds the rate of glucose production and, beyond the newborn period, most commonly presents when an intercurrent illness is associated with reduced calorie consumption caused by anorexia (prolonged fasting), vomiting, and diarrhoea. Hypoglycaemia may also occur after a period of less protracted (e.g. overnight) fasting when blood glucose production is limited by an impaired counter‐regulatory response resulting from cortisol and/or growth hormone deficiency, an inborn error of metabolism affecting glycogenolysis or gluconeogenesis, or when an increased rate of glucose utilization occurs together with decreased hepatic glucose production (hyperinsulinism, malaria, sepsis). The causes of hypoglycaemia in infants and children are shown in Table 2.3. Table 2.3 The differential diagnosis of hypoglycaemia in children. When hypoglycaemia occurs in a neonate, the following details should be obtained: In the older child, clarify the following details: Individuals presenting with hypoglycaemia usually do not have abnormalities on clinical examination. Nonetheless, evaluation of growth (obtain all available growth records), weight gain, and a careful physical examination may identify abnormalities that will inform the cause of the child’s hypoglycaemia. Table 2.4 lists the most common signs and associated disorders (see Figure 2.2). Table 2.4 Clinical signs on examination. Figure 2.2 Ear lobe crease in a child with Beckwith–Wiedemann syndrome. Although hypoglycaemia is a clinical emergency requiring prompt therapy, whenever possible, a blood sample (often referred to as the ‘critical sample’) for investigations should be drawn before administering glucose. Urine should be collected at the earliest opportunity and stored together with blood samples at –20 °C or below. The processing of these samples should be discussed with the laboratory to ensure proper storage of samples. The samples obtained at the time of hypoglycaemia may produce clear biochemical evidence of the cause of the hypoglycaemic episode and thus avoid having to subject the child to further costly and potentially hazardous investigations. Interpretation of the ‘critical’ blood and urine samples obtained at the time of presentation with hypoglycaemia (plasma glucose <50 mg/dL, <2.8 mmol/L), together with a careful physical examination, including assessment of the size of the liver, can frequently determine the specific aetiology of hypoglycaemia (Figure 2.3). Figure 2.3 An algorithm for evaluation of hypoglycaemia. Urine ketones 0 to + indicates absent, trace or small ketonuria; ++ to +++ indicates moderate or large ketonuria; FFA, free fatty acids; BOHB, beta‐hydroxybutyrate; serum insulin concentrations >100 μU ml−1 together with a low C‐peptide level suggests factitious hypoglycaemia. The preliminary investigation of hypoglycaemia is shown in Table 2.5. If blood and urine samples obtained at the time of initial hypoglycaemia do not elucidate its cause (or were not obtained), then additional investigations may be considered. These may include the measurement of intermediary metabolites before and after meals or a prolonged fast performed under controlled conditions, followed by the investigations listed in Table 2.5. Because a prolonged fast may result in severe hypoglycaemia and is potentially dangerous, fasting studies must be performed at a specialist centre. The length of the fast will be determined by the age of the child (Table 2.6). Children undergoing a fasting study should be closely supervised by staff who are experienced in dealing with hypoglycaemia. The timing of the latter stages of the fast when the child is most likely to become hypoglycaemic should be planned to coincide with a time of the day when appropriate staff are available to monitor the patient and assist in any required resuscitation. Intravenous (IV) glucose and hydrocortisone should be available at the bedside. All symptoms should be carefully documented, sample times clearly recorded, and the patient must never be left unsupervised until the fast is complete and the patient is treated with glucose and/or food. Should hypoglycaemia occur during the planned fasting test, the treatment is described below (see section ‘Treatment’). Table 2.5 Investigation of hypoglycaemia at time of presentation. ACTH, adrenocorticotrophic hormone. a Ethanol, salicylates, sulphonylureas. Table 2.6 Length of fasts for children undergoing investigation of possible hypoglycaemia. Investigations may fail to identify a specific cause of hypoglycaemia, which may occur in the context of a normal endocrine counter‐regulatory response, associated with appropriately raised free fatty acids and ketones. Plasma lactate and pyruvate may be normal but alanine low, suggesting a reduced supply of gluconeogenic precursors. This disorder of unknown aetiology is known as ‘accelerated starvation’ or ‘idiopathic ketotic hypoglycaemia’. It is more common in boys than girls and may be associated with a history of foetal growth restriction or a thin physique. Hypoglycaemia may occur after a day of unusually intense physical activity and/or decreased calorie consumption. Episodic hypoglycaemia usually resolves spontaneously by age 7–9 years. There are numerous causes of hypoglycaemia associated with hyperketonaemia (Figure 2.3). Therefore, accelerated starvation or idiopathic ketotic hypoglycaemia should only be diagnosed after other endocrine and metabolic causes of hypoglycaemia have been carefully excluded. The aim is to raise the blood glucose concentration, provide adequate fuel for the brain, and decrease the risk of brain injury. Owing to uncertainty regarding the definition of hypoglycaemia and appropriate thresholds for intervention, and a desire to promote breast‐feeding and not separate infants from their mothers, the most common treatment for asymptomatic hypoglycaemia is to increase the frequency of feeding. Dextrose gel (200 mg/kg) massaged into the buccal mucosa before a feed is effective in raising blood glucose concentrations in hypoglycaemic late pre‐term and term babies in the first 48 hours after birth. In conjunction with breast‐feeding, dextrose gel is a non‐invasive alternative to infant formula as a first‐line treatment for neonatal hypoglycaemia. Babies who remain hypoglycaemic after initial treatment, or whose blood glucose concentrations are extremely low, are admitted to a newborn intensive care unit for treatment with intravenous dextrose (200 mg/kg of 10% dextrose as an IV bolus followed by a continuous infusion at a rate of 8 mg per kg per minute). For high‐risk neonates without a suspected congenital hypoglycaemia disorder, the goal of treatment is to maintain plasma glucose >50 mg/dL [2.8 mmol/L] in infants <48 hours and >60 mg/dL [3.3 mmol/L] >48 hours from delivery. Once the initial (‘critical’) blood samples have been obtained (see Table 2.5), the patient should be given IV dextrose (see section ‘Emergency Management of Acute Hypoglycaemia’). The response to treatment should be monitored and the infusion rate adjusted to maintain blood glucose concentrations >4 mmol/L [72 mg/dL]. The use of intermittent boluses of glucose in concentrations greater than 25% should be avoided because of the risk of cerebral oedema. If the patient remains unconscious despite normalization of the blood glucose concentrations, IV hydrocortisone should be administered empirically for possible undiagnosed adrenal insufficiency. If the patient does not respond to hydrocortisone, the possibility of an intracranial disorder or an inborn error of metabolism should be considered. 1 Obtain initial diagnostic blood sample. 2 Give 0.2 grams glucose per kg body weight (2 ml/kg of 10% dextrose) intravenously over 4 to 6 minutes. 3 Start an infusion of IV 10% dextrose at an initial rate of 10 mg per kg body weight per minute (6 ml/kg/h) and adjust according to blood glucose response. 4 If patient remains unconscious, give 25 mg per metre2 hydrocortisone intravenously. 5 Collect next urine sample for diagnostic investigations. Hyperinsulinism should be considered when glucose requirements to maintain normoglycaemia exceed 8–10 mg per kg body weight per minute (see Table 2.3 and diagnostic criteria in Box 2.1). Persistent hyperinsulinaemic hypoglycaemia, also referred to as congenital hyperinsulinism, is the most common cause of recurrent or persistent hypoglycaemia in infants. To date, mutations of 10 different genes involved in controlling insulin secretion have been described. Inactivating mutations in the genes ABCC8 and KCNJ11, encoding SUR1 and Kir6.2, the two subunits of adenosine triphosphate (ATP)‐sensitive (KATP) channels in the pancreatic beta cells, cause the most common and severe forms of congenital hyperinsulinism. Early recognition, accurate diagnosis, and immediate effective management to maintain normal blood glucose levels (at least 70 mg/dL, 4 mmol/L before feeds) are crucial. Delay in the diagnosis or ineffective management leads to hypoglycaemia‐induced brain injury and adverse neurodevelopmental outcomes.
Hypoglycaemia
Physiology
Premature or postmature delivery
Foetal growth restriction (small for gestational age)
Maternal gestational diabetes; infant of a diabetic mother
Large for gestational age
Perinatal stress associated with transient neonatal hyperinsulinism
Maternal intrapartum treatment with glucose or with sulphonylureas
Abrupt interruption of an infusion with a high glucose concentration
Family history of a genetic form of hypoglycaemia
Congenital syndromes; e.g. Beckwith‐Wiedemann or abnormal physical features (midline facial defects, micropenis, cryptorchidism) suggestive of hypopituitarism
Symptoms and signs of hypoglycaemia
Neurogenic (autonomic)
Neuroglycopenic
Jitteriness/tremors
Poor suck or feeding
Sweating
Weak or high‐pitched cry
Irritability
Hypotonia
Tachypnoea
Change in level of consciousness (lethargy, coma)
Pallor
Seizures
Definition
Aetiology
Accelerated starvation (idiopathic ketotic hypoglycaemia)
Hyperinsulinemic hypoglycaemia
Hormone deficiency
Glycogenoses
Glucose transporter defect
Disorders of gluconeogenesis
Disorders of fatty acid oxidation
Disorders of ketogenesis
Drugs (insulin, sulphonylureas, ß‐blockers, aspirin, alcohol)
Infections (malaria, septicemia, gastroenteritis)
Preliminary examination and investigation
History and physical examination
Clinical sign
Possible diagnosis
Optic atrophy
Septo‐optic dysplasia
Cranial midline defects
Growth hormone deficiency
Short stature or decreased height velocity
Growth hormone deficiency
Micropenis
Growth hormone deficiency
Diffusely increased skin or buccal pigmentation
Addison’s disease
Hypotension
Addison’s disease
Underweight or malnutrition
Accelerated starvation
Tall stature or increased height velocity
Hyperinsulinism
Excess weight
Hyperinsulinism
Abnormal ear lobe creases (Figure 2.2)
Beckwith–Wiedemann syndrome
Macroglossia
Beckwith–Wiedemann syndrome
Umbilical hernia
Beckwith–Wiedemann syndrome
Hemihypertrophy
Beckwith–Wiedemann syndrome
Hepatomegaly
Glycogen storage disorder 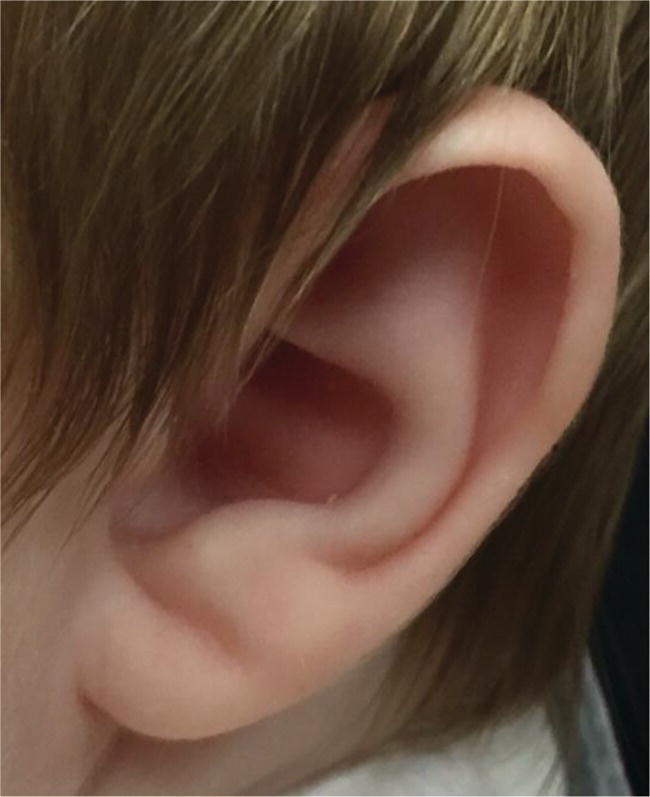
Investigations
Sample
Investigation
Blood sample
Glucose
Bicarbonate or venous pH
Free fatty acids and β‐hydroxybutyrate
Lactate
Insulin and C‐peptide
Cortisol and ACTH
Growth hormone
Liver function tests
Ammonia
Urea and electrolytes
Alanine
Acylcarnitines; total and free carnitine
Administer glucose
Next urine sample
Ketones
Glucose and reducing sugars
Organic acid profile
Glycine conjugates
Carnitine derivatives
Toxicology screena
Age
Duration (hr)
<6 mo
8
6–8 mo
12
8–12 mo
16
1–2 yr
18
2–4 yr
20
4–7 yr
20
>7 yr
24
Treatment
Neonatal hypoglycaemia
Acute treatment at initial presentation in older children
Emergency management of acute hypoglycaemia
Hyperinsulinism
Medical treatment
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


