Abbreviation: NRTI = nucleoside and nucleotide reverse transcriptase inhibitors; TAM = thymidine analog mutation; GFR = glomerular filtration rate.
| Agent (trade name in italics) | Metabolism | Dosing | Resistance | Adverse effects | Miscellaneous |
|---|---|---|---|---|---|
| Nevirapine (NVP) Viramune | Hepatic CYP3A4, CYP2B6 | 200 mg PO qd for 2 weeks, then 400 mg PO qd Adjustment needed for dialysis patients | K103N V106A/M Y181C Y188L G190A/S | Rash, including Stevens–Johnson syndrome Hepatic necrosis | First-generation NNRTI Preferred NNRTI in pregnancy Contraindicated in women with baseline CD4 >250 cells/mm3 and men with CD4 >400 cells/mm3 owing to risk of hepatic toxicity Can precipitate acute methadone withdrawal |
| Efavirenz (EFV) Sustiva | Hepatic CYP3A4, CYP2B6 | 600 mg PO qd on empty stomach | K103N 100 106 181 188 190 225 | Rash Hepatotoxicity Vivid dreams Insomnia Dizziness Poor concentration Teratogenicity (Pregnancy Category D) | Current preferred NNRTI First-generation NNRTI Must be taken on empty stomach (usually before bed) to minimize side effects Can precipitate acute methadone withdrawal Pregnancy Category D |
| Etravirine (ETR) Intelence | Hepatic CYP3A4, CYP2C9, CYP2C19 | 200 mg PO q12h Absorption decreased with food 50% | Y181C G190A Multiple other mutations | Rash Nausea | Second-generation NNRTI Only medication of this class that cannot be inactivated by a single point mutation |
| Rilpivirine (RPV) Edurant | Hepatic CYP3A4 | 25 mg PO qd, taken with minimum 400-kcal meal Caution in severe renal impairment | V90I K101E/P/T E138K/G V179I/L Y181I/C V189I H221I F227C/L M230L | Depression Insomnia Headache Rash QT prolongation | Current alternative NNRTI Second-generation NNRTI Take with minimum 400-kcal meal Contraindicated with PPIs, anticonvulsants, rifampin/rifabutin/rifapentine, dexamethasone, St. John’s wort (all decrease RPV levels) H2Bs must be taken 12 h before or 4 h after RPV Antacids should be taken 2 h before or 4 h after RPV Caution with combination with medications that prolong QT interval Contraindicated when initial HIV viral load >100 000 copies/mL (more virologic failure than efavirenz) |
Abbreviations: NNRTI = non-nucleoside reverse transcriptase inhibitor; PPI = proton pump inhibitor; H2B = H2 histamine receptor blocker.
Abbreviations: PI = protease inhibitor; PPI = proton pump inhibitor; H2B = H2 histamine receptor blocker.
HIV replication cycle
A better understanding of the HIV replication cycle (Figure 99.1) has led to the development of antiretroviral medications targeted against viral enzymes and even host proteins. A visualization of this replication cycle is essential to understanding how these medications function. Current ART medications inhibit entry of virus into host cells, reverse-transcription of viral DNA from an RNA template, integration of this viral DNA into the host genome, and processing of newly transcribed viral proteins.
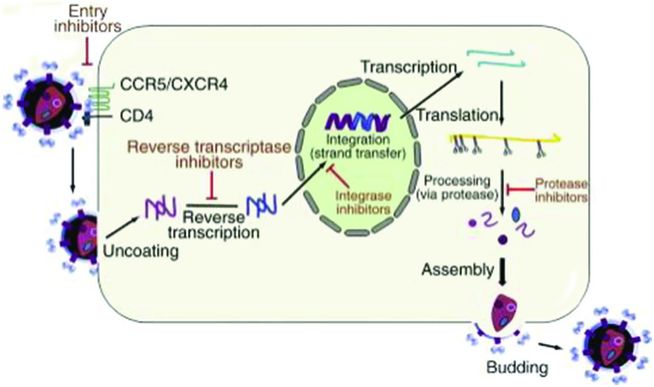
Figure 99.1 HIV replication cycle.
Fusion of HIV with the host cell membrane with subsequent viral RNA entry into the host cell is the first step of HIV viral replication (Figure 99.1). Viral glycoproteins 120 and 41 group together and interact with host cell CD4 receptors and CCR5 or CXCR4 co-receptors, causing fusion of the viral and host membranes and entry of viral factors (Figure 99.2).
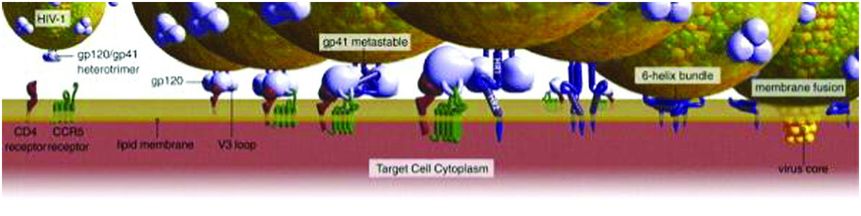
Figure 99.2 Fusion of the viral and host membranes.
After fusion, viral RNA is released into the host cell along with essential viral enzymes, including the viral reverse transcriptase enzyme (RT). RT then uses host cell nucleosides and nucleotides to construct a double-stranded viral complementary DNA (cDNA). Viral cDNA interacts with the viral integrase protein in the host cell cytoplasm. Viral cDNA is then transported into the nucleus of the host cell, where integrase incorporates viral cDNA into the host DNA genome. Once incorporated into the host genome, viral DNA is transcribed and translated into polyproteins by host enzymes and ribosomes. Viral proteases then cleave these polyproteins into functional and mature viral proteins. Full HIV virions are constructed. These new virions then bud from the host cell surface, detach, and infect new host cells, repeating the cycle (Figure 99.1).
HIV antiretroviral drug classifications
Nucleoside and nucleotide reverse transcriptase inhibitors (NRTIs)
Viral RT uses a pool of available host cellular nucleosides and nucleotides to construct viral cDNA from a viral RNA template. NRTIs are designed to mimic the structure of these natural nucleosides and nucleotides. When viral RT erroneously incorporates an NRTI into the growing viral cDNA chain, the chain is prematurely terminated. NRTIs block further chain elongation in the 3′ direction. Table 99.1 presents a summary of the drugs in the NRTI class.
With few exceptions, two NRTI agents are combined to form an “NRTI backbone” that comprises two of the three active drugs in a modern ART regimen. A third active agent is then added to this NRTI backbone.
Emtricitabine (FTC) and lamivudine (3TC) are similar, roughly equivalent cytosine analogs, and are both very well tolerated. One of these two medications is typically combined with a second preferred, alternative, or third-line agent of this class to form most front-line NRTI backbones. They have the additional advantage of being able to be dosed once daily.
Tenofovir disoproxil fumarate (TDF) is currently the preferred NRTI and is generally used in combination with FTC. TDF is extremely well tolerated with regard to side effects, can be given once daily, and has activity versus hepatitis B virus. It has the advantage of once-daily dosing. Its major disadvantages are a potential for renal dysfunction and osteoporosis with long-term use. It must be adjusted for renal function and is not recommended for glomerular filtration rate (GFR) <30 mL/min (Table 99.1).
Abacavir (ABC) is the main alternative NRTI and is generally combined with 3TC. Like TDF, it is also extremely well tolerated by patients and is given once daily. Some data have suggested a possible increase in risk of myocardial infarction using ABC, and some conflicting data exist suggesting that ABC may not be as effective in patients with higher baseline viral loads (discussed later). The primary disadvantage of ABC, however, is a severe, life-threatening hypersensitivity reaction that can occur in patients positive for the HLA-B5701 allele. ABC should not be administered to patients positive for the HLA-B5701 allele.
Zidovudine (AZT) is still occasionally used as a third-line NRTI, generally in combination with 3TC. It is poorly tolerated compared with the other commonly used NRTIs previously mentioned, and side effects such as lipodystrophy, peripheral neuropathy, myopathy, anemia, and hepatic steatosis can occur (Table 99.1). An additional disadvantage with AZT is that it must be administered twice daily.
Resistance to NRTIs generally occurs due to RT mutations. Mutations resulting in resistance to NRTIs and to all classes of ART medications occur most commonly in the setting of incomplete adherence to a full ART regimen. Prolonged viremia is often necessary for the development of such mutations. With the exception of the M184V and K65R mutations, significant resistance to a drug may take several serial mutations, which may require several months of viremia.
Table 99.1 contains a listing of specific mutations conferring resistance to each drug in the NRTI class, but there are several major mutations of notable importance. Thymidine analog mutations (TAMs) were commonly seen prior to the use of triple-drug therapy, especially in patients taking AZT or stavudine (d4T). They allow for excision of NRTIs from the growing viral cDNA strand. TAM1 mutations occur at positions 41, 210, and 215 on RT and can confer resistance to multiple drugs across the NRTI class, including newer NRTIs such as TDF (for example, TAMs can decrease TDF activity by a factor of 4). TAM2 mutations cluster at RT positions 67, 70, and 219 and do not confer class resistance to the extent that TAM1 mutations do. Insertion mutations can be significant as well, with an insertion at position 69 conferring class-wide resistance. Important substitution mutations include the K65R mutation, which can decrease TDF activity as well as the activity of other NRTIs (excluding AZT), and the M184V substitution mutation, which may reduce FTC and 3TC activity by a factor of 1000.
The M184V mutation does confer a fitness disadvantage to the HIV virus and reduces the viral load. M184V also renders HIV highly sensitive to AZT, even in the face of TAM mutations. Therefore, FTC or 3TC therapy may be continued in patients with known M184V mutation as an inactive drug to exploit such secondary advantages. This strategy may prove especially useful in patients with multidrug-resistant (MDR) HIV strains.
Certain general toxicities of this drug class can be attributed to the mechanism of action of NRTIs. In addition to terminating viral DNA transcription, they can also inhibit host DNA transcription enzymes. Mitochondrial DNA polymerase is particularly susceptible to these drugs, and lactic acidosis, peripheral neuropathy, hepatic steatosis, myopathy, and lipoatrophy can all result from inhibition of this enzyme (see description of AZT). Insulin resistance and diabetes may also be potential adverse effects. The first-line NRTIs have minimal overlapping interaction with mitochondrial DNA polymerase, and therefore are much better tolerated than their predecessors.
Non-nucleoside reverse transcriptase inhibitors (NNRTIs)
NNRTIs are allosteric RT inhibitors that bind to a dedicated site that lies apart from the enzyme’s active functional site. The result of NNRTI binding is a change to the enzyme’s conformation, rendering it ineffective at viral DNA transcription. Table 99.2 presents a summary of the drugs in the NNRTI class.
First-generation NNRTIs include nevirapine (NVP) and efavirenz (EFV). EFV is considered the preferred medication of this class. It has demonstrated superior virologic suppression compared with triple-NRTI regimens or protease inhibitor (PI)-based regimens with nelfinavir (NFV), indinavir (IDV), and ritonavir-boosted lopinavir (LPV/r) and noninferior suppression compared with atazanavir-based (ATV) therapy. EFV can be administered once daily. EFV should be taken on an empty stomach to minimize side effects, which can be substantial. Neuropsychiatric side effects such as vivid dreams and fogginess have been described in approximately 50% of patients, but are generally mild and self-limited (within 2 to 6 weeks). EFV is the only US Food and Drug Administration (FDA) pregnancy category D antiretroviral and should not be initiated during pregnancy. NVP should not be administered to men with a CD4 count >400 cells/mm3 and women with a CD4 count >250 cells/mm3, as usage in these groups has been associated with hepatic necrosis.
Second-generation NNRTIs include etravirine (ETV) and rilpivirine (RPV). These NNRTIs have been chemically altered from their predecessors with the theoretical goal of rendering them more effective against drug-resistant HIV. RPV, the newest drug from the NNRTI class, has been demonstrated to have a higher rate of virologic failure compared with EFV in patients with higher HIV viral loads, thus earning it a designation as an alternative NNRTI. The E138K mutation is commonly seen in the setting of RPV virologic failure, and the M184V/I and K65R/N mutations are seen with greater frequency than with EFV in this setting. Administration of RPV in patients with a viral load >100 000 copies/mL should be avoided, and its use is only approved in treatment-naïve patients. RPV must also be taken with a high-calorie meal. Its absorption requires an acidic environment, so care must be taken to avoid direct coadministration with antacids and H2-blocking medications. Proton pump inhibitors should be avoided altogether in patients on RPV. RPV may cause an artifactual rise in creatinine that does not reflect a true change in the GFR.
ETV has only been studied in treatment-experienced patients and is approved only in this setting (see ART selection in patients with multidrug-resistant HIV, below).
Unfortunately, since it is not directly involved in the action of the RT enzyme, the conformation of the dedicated NNRTI binding site is not important for RT functionality. Therefore, mutations at this site may block NNRTIs from binding, but generally do not affect RT functionality. Simple single-point mutations to the enzyme can render every first-generation NNRTI ineffective (hence the development of second-generation NNRTIs mentioned previously). Currently, ETV is the only NNRTI that is approved in the setting of NNRTI mutations. The low barrier to resistance with the NNRTIs may limit the use of these medications in patients with a history of poor compliance to ART.
NNRTIs are hepatically metabolized by cytochrome P450 (CYP) enzymes, making drug–drug interactions a concern. Particular care must be taken when combining these medications with anticonvulsant, antimycobacterial, and certain antifungal therapies. PIs may have strong interactions with NNRTIs as well, and their combination should generally be avoided, except in special cases.
Protease inhibitors (PIs)
PIs inhibit HIV aspartyl protease, an enzyme used by the virus to cleave and construct gag and pol proteins. Gag proteins form structural aspects of the virus, while pol proteins comprise the vital viral enzymes RT, protease, and integrase. PIs are primarily metabolized by hepatic CYP enzymes, especially CYP3A4. They may also induce or inhibit these enzymes. Drug–drug interactions are therefore significant, especially in the setting of coadministration with immunosuppressants, antiarrhythmics, antibiotics, statins, opiates, oral contraceptives, benzodiazepines, or other ART medications. For example, acute withdrawal may occur when PIs are administered to patients taking chronic opiates such as methadone. Efficacy of contraceptives may be reduced significantly. Table 99.3 presents a summary of the drugs in the PI class.
The metabolism of PIs by CYP3A4 is exploited with the concept of “boosting.” The PI ritonavir (RTV) is an especially powerful inhibitor of CYP3A4. Owing to pill burden, toxicity, and a host of potential drug–drug interactions via its effect on other CYP enzymes, RTV is no longer considered a viable stand-alone PI. However, when RTV is given at subtherapeutic doses in combination with another “primary” active PI, CYP3A4 metabolism becomes saturated, allowing plasma concentrations of the primary PI to become higher (“boosted”) and more stable (increased half-life) with less frequent dosing. The overall effect of this strategy is that fewer pills are required to be taken on a less frequent basis with less toxicity to the patient. This boosting effect also minimizes viral protease mutations by keeping the steady state of drug well above the minimum inhibitory concentration required to suppress HIV replication, even in the face of occasional missed doses. However, while boosting does allow for higher fidelity concentrations of less toxic drugs, adverse effects and drug–drug interactions still do exist and may become accentuated as well. RTV toxicity is also limited at this subtherapeutic dose, yet some effects such as nausea and vomiting may become clinically relevant.
Currently, darunavir (DRV) and atazanavir (ATV) are the preferred medications of the PI class, both boosted by RTV. Each has the advantage of once-daily dosing in treatment-naïve patients. ATV was found to be noninferior with respect to virologic response compared with EFV. Boosting with RTV was clearly shown to be effective when RTV-boosted ATV (ATV/r) was compared with unboosted ATV and with LPV/r. Once-daily DRV/r was demonstrated to be noninferior to LPV/r in treatment-naïve patients. DRV has also shown to be an effective choice in treatment-experienced and MDR HIV, although it must be given twice daily if any DRV mutations exist (see section below on selection of PI and MDR HIV).
Dyslipidemia is a toxicity seen with drugs from this class (ATV having the least effect in this regard). Propensity toward insulin resistance and diabetes may potentially occur as well. Gastrointestinal symptoms are well documented, including abdominal pain, nausea, vomiting, and diarrhea. With regard to the primary preferred PIs, ATV has the notable effect of causing reversible indirect hyperbilirubinemia due to inhibition of UDP-glucuronosyltransferase. Jaundice or scleral icterus may even become evident in patients taking ATV, and a change to an alternative agent should be considered if cosmetic concerns exist. While isolated hyperbilirubinemia itself does not constitute hepatic failure in this setting, the presence of elevated hepatic transaminases should raise concern for alternative pathology.
As mentioned previously, newer boosted PIs have a high barrier to viral resistance. Several mutations to viral protease are usually required for resistance, and generally one or more mutations must be major. DRV is especially robust in the setting of multiple PI mutations.
Entry inhibitors
Entry inhibitors are drugs targeted at inhibition of HIV fusion and entry into host cells. The primary drugs of this class are enfuvirtide (ENF) and maraviroc (MVC). Neither of these medications is currently considered amongst the preferred ART medications.
ENF is an amino acid polypeptide fusion inhibitor and is used primarily as a salvage drug for patients who have prior experience with ART and multiple resistance mutations to primary ART medications. Two randomized trials with median patient CD4 count <100 cells/mm3 demonstrated significantly improved viral suppression when ENF was added to an optimized ART regimen. ENF is dosed as a 90-mg subcutaneous injection administered twice daily. Advantages of this drug include no need to adjust dose for renal or hepatic function and no known drug interactions. There is also no evidence of teratogenicity in animal models (although experience in pregnant humans is lacking). A major disadvantage, however, is the need for twice-daily injections to avoid gastrointestinal denaturation of the medication. Not only is the need for injection a potential psychological barrier to home, but there is also a 98% incidence of local injection reactions. Injections must be administered away from recent previous injection sites or areas of reactive skin, and care must also be taken to avoid blood vessels, large nerves, injured skin, and tattoos. Nausea, vomiting, diarrhea, fatigue, and insomnia are additional adverse effects that have been described. Viral resistance can occur with amino acid substitutions on viral glycoprotein 41, usually at position 36, 38, 40, and 43.
MVC is a CCR5 allosteric antagonist. It is the only approved ART medication that targets a host, not a viral, receptor. It is designed to prevent interaction of viral gp120 with host CCR5, thereby inhibiting viral attachment. It has been demonstrated to effectively lower viral load and raise CD4 counts in two randomized, double-blind, placebo-controlled trials. Patients must be screened with a tropism test prior to initiation to ensure their specific HIV strain does not use CXCR4 for entry, either in lieu of or in conjunction with reliance on CCR5, as MVC has not been shown to be of benefit in this patient population. MVC was shown to be most effective in these two studies when combined with at least two other effective antiretroviral medications. If used in combination with ENF for salvage regimens, patients responded best when there was no ENF resistance and when ENF was being administered to the patient for the first time. The 48-week MERIT study in which MVC plus AZT/3TC was compared with EFV plus AZT/3TC (a standard primary initial HAART regimen at the time of that study) failed to demonstrate that the MVC regimen conferred as much viral suppression as the EFV regimen. However, reanalysis of these data in the MERIT-ES study with a newer Trofile test demonstrated improved efficacy. MVC can be used in combination with two NRTIs in special circumstances for treatment-naïve patients, although it is considered to be a less satisfactory regimen compared to preferred or alternative NNRTI-, PI-, or integrase inhibitor-based regimens.
MVC is dosed at 300 mg twice daily. There is little data to date to suggest dose adjustment in patients with renal or hepatic dysfunction. MVC is metabolized by CYP3A, and therefore dose adjustment is necessary with CYP3A inducers or inhibitors. Concurrent medications that inhibit CYP3A that may be encountered in the HIV population include PIs and clarithromycin, while common inducers encountered in this population include EFV, NVP, and anticonvulsants. Dosing should be decreased to 150 mg twice daily in the presence of an inhibitor, and increased to 600 mg twice daily with an inducer. Side effects of MVC include dizziness, cough, rash, upper respiratory tract infection, and fever. There are two major pathways of viral resistance to MVC. The first occurs with an amino acid substitution on viral gp120. The second, and much more common, pattern of resistance occurs when selection of CXCR4-binding virus occurs (patients initially screened for CXCR4-using virus may have low levels present that are undetectable by the lab assay), as was demonstrated in the MOTIVATE trials.
Aside from drug resistance, selection of CXCR4 virus in patients prescribed MVC may have additional consequences. Patients with CXCR4-predominate virus may develop a faster drop in CD4 cell count and more aggressive progression to AIDS. Discontinuation of MVC in patients who develop CXCR4 tropism may lead to a resurgence of CCR5-predominate infection in such patients (however, MVC should not be reconsidered as a viable therapeutic option in such patients, even if CCR5-predominate virus returns).
Integrase strand transfer inhibitors (INSTIs)
INSTIs are a class of antiretroviral medications designed to block HIV integrase-mediated incorporation of HIV cDNA into the host DNA genome.
Raltegravir (RAL) was the first drug developed in this class and is considered the preferred INSTI. It has been demonstrated to be noninferior and better tolerated than EFV-containing primary ART regimens for treatment-naïve patients. RAL was also shown to decrease the viral load faster within 24 weeks compared to EFV-based regimens (the clinical significance of this class-wide INSTI effect is unknown).
RAL also has demonstrated efficacy in treatment-experienced patients. It showed greater suppression of HIV viral load at 48 weeks compared to placebo when added to an optimal ART backbone in two randomized, double-blinded phase III studies. Of note, in one of these studies patients with resistant virus concurrently started on DRV and ENF for the first time had even greater response, although the study was not powered to assess this effect. In another investigation of RAL use in treatment-experienced patients, substituting RAL for ENF maintained virologic suppression.
Despite its utility in treatment-experienced patients, RAL has a low barrier to resistance. Primary RAL resistance mutations to the viral integrase protein exist as substitutions at Y143, Q148, and N155 on the integrase enzyme.
RAL is administered orally at 400 mg twice daily. Dose adjustment is not necessary in renal or hepatic insufficiency, unless hepatic insufficiency is severe. It is metabolized by glucuronidation and therefore has few drug–drug interactions. RAL should be administered as 800 mg twice daily if coadministered with rifampin.
Elvitegravir (EVG) is the second INSTI to be developed. It is currently approved only for treatment-naïve patients as part of a four-drug fixed-dose combination pill trade (EVG/COBI/TDF/FTC) and is considered an alternative INSTI. This regimen contains 150 mg of EVG, 150 mg of cobicistat
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


