Chapter Outline
NON–LANGERHANS CELL HISTIOCYTOSES
HEMOPHAGOCYTIC LYMPHOHISTIOCYTOSIS
The histiocytoses constitute a collection of rare hematologic diseases that resist easy classification, at least in part because of the imprecise definition of “histiocyte.” “Histiocyte” broadly refers both to cells of the macrophage lineage and to dendritic cells (DCs), only some types of which are derived from macrophages. The numerous descriptive and functional subsets of macrophages and DCs only add to the confusion. Nonetheless, their collective consideration is justified by a degree of uniformity in these childhood diseases that present with infiltration of bone, secondary lymphoid organs, or the liver, with or without concomitant involvement of other visceral organs. However, prognosis and treatment options are specific to the subtype, and thus an appreciation of the individual varieties of histiocytoses is mandatory.
This chapter is divided into two main sections that correspond to the two most common clinical histiocytic disorders, Langerhans cell histiocytosis (LCH) and hemophagocytic lymphohistiocytosis (HLH). Also included is a relatively brief discussion of non–Langerhans cell histiocytoses and Rosai-Dorfman disease. Each section will begin with a description of the normal counterpart(s) of the relevant histiocytes and their ontogeny to provide a context for describing how these cells are pathologically involved in the histiocytic disorders. Each section will also include a description of their clinical manifestations and current approaches to treatment.
Types of Dendritic Cells
The pathologic cells of LCH share some features with normal epidermal Langerhans cells (LCs), which are the primary antigen-presenting cells (APCs) of skin. LCs are one type of the general class of APCs known as DCs because of the characteristic dendritelike structure they assume when activated. DCs are, in turn, just one of several types of APCs that are called “professional” because they can, by themselves, fully activate naive T lymphocytes by providing both the antigen/major histocompatibility complex ligand for T-cell receptor binding and the accessory signals required for full activation.
Because DCs can also render T cells tolerant to specific antigens, they have become a major focus of basic immunologic research. This research has revealed a surprising level of functional and phenotypic diversity among DCs, which will undoubtedly be relevant to the proliferative disorders affecting these cells. Several authors have provided helpful guides to thinking about and classifying DCs. Overall, DCs can be divided into conventional and inflammatory types ( Box 64-1 ) . Conventional DCs are present in lymphoid and nonlymphoid organs in their basal state, and their primary function is to collect antigens for presentation to T cells in order to activate or tolerize them, depending on the antigen’s source (i.e., nonself vs. self). Inflammatory DCs arise in response to specific inflammatory or infectious signals and are ordinarily not identifiable in the nonchallenged state. In addition to presenting antigen, these cells secrete cytokines and other mediators, including tumor necrosis factor (TNF), that enhance host defense.
Conventional Dendritic Cells
Migratory Dendritic Cells
Migratory dendritic cells (DCs) gather antigen in peripheral tissues, then migrate to regional lymph nodes to present antigen to T lymphocytes
Examples: Langerhans cells, dermal DCs, interstitial DCs
Lymphoid Tissue Dendritic Cells
Lymphoid tissue DCs gather antigen within lymphoid tissue and present antigen to T lymphocytes within the same lymphoid tissue
Examples: thymic DCs, splenic DCs
Inflammatory Dendritic Cells
Inflammatory DCs arise in response to inflammatory signals; they can gather antigen and present antigen to T lymphocytes and can secrete cytokines
Example: tumor necrosis factor and inducible nitric oxide synthase–producing DCs
Mucosal Dendritic Cells
Mucosal DCs are resident in mucosal surface tissues; they can gather antigen and present antigen to T lymphocytes and help direct responses toward activation or tolerance
Predendritic Cells
Predendritic cells can develop DC function directly in response to inflammatory stimuli
Example: plasmacytoid DCs, monocytes
Within the conventional DC category, one can distinguish between migratory DCs and DCs that reside in lymphoid tissue. Migratory DCs fulfill the sentinel role of this leukocyte class and include LCs, dermal DCs, and interstitial DCs of other organs. Migratory DCs shuttle between end organs that interface with the external world (e.g., skin or mucosa), in the case of LCs, and regional lymph nodes. This trafficking occurs at a low level in the basal state but can be greatly enhanced in the presence of foreign antigen or inflammatory challenges. The migratory DC takes up antigen in the periphery and, once activated, travels to regional nodes either to present antigen itself or to transfer antigen to resident DCs in the node, which then perform the presentation function to T cells. Although the molecular signals that control this migratory behavior are not understood in detail, chemokines and their receptors play a large role. In the case of LCs, resting cells express the chemokine receptor CCR6 whose ligand, CCL20, is secreted by cutaneous keratinocytes. Upon antigen uptake and activation, LCs downregulate CCR6 and in its place upregulate another chemokine receptor, CCR7, whose ligands, CCL19 and CCL21, are secreted by cells in regional lymph nodes. This receptor switch has the dual effect of neutralizing the LC’s anchor to the skin and attracting them to regional lymph nodes.
In contrast, DCs that reside in lymphoid tissue are nonmigratory. They make up most of the DCs populating the thymus and spleen and about half of the DCs in lymph nodes. Unlike migratory DCs, which are mature when they appear in lymph nodes, resident DCs are immature, which allows them to take up, process, and present local antigens. Surface markers can distinguish several subsets of resident DCs, which are presumed to have specialized functions. In the mouse, these subsets include cluster of differentiation (CD)8+ and CD8− cells, and among the CD8− population are CD4+ and CD4− subpopulations. Specific DC subsets also have characteristic gene expression profiles.
LCs bear distinctive intracellular and extracellular markers that permit their distinction from DCs that reside in lymphoid tissue. The most characteristic surface marker is CD1a, and the most characteristic ultrastructural feature is Birbeck granules, which are pentilaminar “tennis racket”–shaped cytoplasmic organelles that appear to be uniquely present in LCs. Langerin, also known as CD207, is a cell surface marker for LCs that associates with Birbeck granules when internalized. Langerin is a C-type lectin with specificity for sugars that contain mannose, suggesting that it may be involved in processing and trafficking of antigens that bear these sugars. Dectin-2 is another C-type lectin restricted to DCs, and DEC-205 (CD205) is yet another lectin that is often used to identify LCs, although its expression is not as restricted to LCs as are the other lectins. In the appropriate histologic context, from a diagnostic standpoint, CD1a and Langerin are the most reliable lineage-specific markers.
Some authors also distinguish a group of so-called “predendritic cells” that do not ordinarily have DC functions or structure. However, these cells can develop directly into DCs upon stimulation without the need for proliferation. One well-studied example is the plasmacytoid DC, which circulates as a nondescript mononuclear cell but acquires APC function and secretes large amounts of alpha interferon when activated. Technically, using this definition, monocytes would also be predendritic cells. After exposure to interleukin (IL)-4 and granulocyte-macrophage colony-stimulating factor (CSF), monocytes become functional DCs, although their phenotype is closer to inflammatory DCs than conventional DCs.
Origins of Dendritic Cells
The availability of genetically modified mice that carry lineage-restricted markers has greatly expanded our understanding of the origins and development of DCs. Although mouse models have some limitations, the major insight these animals provide is that a salient characteristic of DC ontogeny is flexibility. For example, although DCs generally have myeloid characteristics, all DC subtypes in the mouse can arise from either committed lymphoid or committed myeloid precursors. Even though half of thymic DCs show evidence for immunoglobulin rearrangements, suggesting a lymphoid phenotype, they also transcribe the macrophage colony-stimulating factor (M-CSF) receptor gene, which is a myeloid characteristic. Activation of fms-like tyrosine kinase–3 (FLT3), a growth factor receptor, is required for DC development, and it has been suggested that committed myeloid or lymphoid precursors expressing FLT3 can differentiate into DCs in the presence of FLT3 ligand.
Splenic DCs have a relatively rapid turnover rate (every 3 days). These cells are repopulated both by DC division and splenic precursors, as well as by bone marrow precursors. In contrast, LCs are very long-lived. After their migration from the skin, the population is replenished from a pool of LC precursors in the skin, as well as by circulating monocytes. The latter is dependent on the action of the chemokine CCL2 acting on its receptor CCR2. Thus the ontogeny of LCs is distinct from that of splenic DCs. Immature DCs are more closely related to LCs but appear to be derived from noninflammatory monocytes that do not express CCR2. Plasmacytoid DCs split off from conventional DCs early in development, consistent with their distinct functional characteristics.
Langerhans Cell Histiocytosis
The hallmark of LCH is the accumulation of LC-like DCs in one or more tissues or organs. The clinical manifestations of LCH are highly diverse. They result both from the direct, local effects of the growth and accumulation of pathologic LCs and the indirect, secondary effects these activated cells have on normal tissues, particularly cells of the immune system. Although the most commonly involved sites of disease are the bone and skin, virtually any organ or system may be involved. The pattern of involvement is not necessarily related to patterns of normal LC or DC migration.
Although the histiocytes in LCH share certain characteristics with LCs, such as expression of CD1a and the presence of Birbeck granules, recent reports suggest that mature LCs may not be the cell of origin for LCH histiocytes. In particular, a global analysis of gene expression suggested that LCH cells are more closely related to myeloid dendritic precursors than to LCs. Whether this pattern of gene expression reflects a non-LC cell origin for LCH or is a consequence of LC transformation remains to be determined.
The fact that pathologic LCs are related to immunomodulatory cells and that they elicit inflammatory infiltrates suggests that LCH might be a reactive rather than a neoplastic disease. Indeed, ample precedent for this mechanism exists among the histiocytoses, including the secondary HLH syndromes that arise in the context of viral infection. However, no reproducible reports exist of viral genomes recovered from LCH cells, and epidemiologic studies are not consistent with an infectious or environmental cause of LCH. Rather, the preponderance of evidence indicates that LCH arises as a consequence of intrinsic genetic abnormalities. The most salient arguments that LCH is a neoplasm and not a reactive disease are as follows: (1) pathologic LCs are clonal, as demonstrated by nonrandom X chromosome inactivation both in whole LCH tissue (in a proportion corresponding to the proportion of CD1a+ cells in the lesion) and in sorted CD1a+ cells, and (2) nearly 60% of LCH samples carry the oncogenic BRAF V600E variant. Notably, the MAPK pathway, which would be activated constitutively in the presence of BRAF V600E, is uniformly activated in LCH cells regardless of the presence or absence of BRAF V600E, suggesting that alternative mechanisms of pathway activation are present in LCH samples that lack BRAF V600E. In some cases, other activating mutations of BRAF have been found. Additional known mechanisms of BRAF activation, such as gene amplification or translocation, which occur in other diseases, have not been described in LCH. Notably, although pulmonary LCH in adults is generally considered to be a polyclonal disease, more than 40% of pulmonary LCH cases contain BRAF V600E. This finding suggests that a significant proportion of these cases are clonal or that multiple independently developed clones of pulmonary LCH have BRAF V600E, giving the overall appearance of polyclonality.
Although BRAF mutations are among the most common molecular abnormalities found in all cancers, they are not present in other histiocytoses that affect children, such as juvenile xanthogranuloma (JXG) or Rosai-Dorfman disease. However, BRAF V600E mutations are present in the histiocytes of about 50% of patients with Erdheim-Chester disease, a rare and aggressive non-LCH of adults. Furthermore, treatment of patients who have BRAF V600E–positive Erdheim-Chester disease with a BRAF inhibitor led to dramatic clinical responses, indicating the pathogenetic importance of this mutation. Interestingly, some of these patients had concomitant LCH that also responded to BRAF inhibition, suggesting that this therapeutic approach may be effective in patients who have BRAF V600E–positive LCH as well.
Activating mutations of BRAF are insufficient by themselves to produce neoplastic disease, and therefore additional molecular alterations are likely to be required for the development of LCH. One of the most common molecular abnormalities in LCH is overexpression of TP53 , suggesting that mutational inactivation of this pathway may contribute to pathogenesis. More such changes in other genes are likely to be discovered as advanced genomic analyses are applied to LCH samples.
Incidence
The incidence of LCH is difficult to determine precisely because of the rarity and marked clinical variability of the disorder. Estimates are in the range of 2.6 to 8.9 cases per million per year for children younger than 15 years, corresponding to roughly one tenth the incidence of acute leukemia in childhood. LCH occurs in people of all races and all ages. Although the peak age at diagnosis is 2 years, LCH can present at any time from birth to old age. No evidence of seasonal variation has been noted in the time of presentation of LCH. Several studies have shown a slight male predominance.
No known predisposing factors exist for the development of LCH in the majority of cases. Studies have demonstrated concordance of the disorder in monozygotic twins, suggesting an inherited predisposition to LCH that might account for ~1% of cases. However, a positive family history is lacking in most cases. Interestingly, isolated pulmonary LCH in adults is closely linked to cigarette smoking, and it often resolves with smoking cessation. However, exposure to cigarette smoke has not been linked to pediatric LCH. As previously noted, a significant proportion of pulmonary LCH cases also carry the activated BRAF V600E allele.
An intriguing (although incompletely understood) association exists between malignancy and LCH. Children with a history of cancer have an increased risk of LCH. Conversely, children with a history of LCH appear to have an increased risk of cancer. This finding suggests the possibility that underlying genetic abnormalities may place individual patients at increased risk for LCH. Alternatively, the association between LCH and cancer may fall within the spectrum of secondary posttherapy effects, because irradiation and drugs such as etoposide are used to treat LCH and are known to induce tumor-promoting genotoxic injury. Several cases of LCH arising in the setting of T-cell acute lymphoblastic leukemia have been described, typically when the leukemia is in remission while the patient is receiving therapy or in the early posttherapy period. A clonal link between the initial leukemia and pathologic LCs of LCH has been established in some cases, suggesting that the LCH represents a manifestation of the malignant clone rather than a distinct reactive or neoplastic process related to the leukemia or its therapy. An intriguing link may exist to hairy cell leukemia, which also harbors BRAF V600E in the majority of cases.
Clinical Features
The clinical manifestations of LCH are protean. Although patterns of clinical presentation have been described, the disease is appropriately viewed as a spectrum. LCH may involve a single site, multiple sites in a single organ system, or multiple organ systems. Many patients present with localized pain, soft tissue swelling, or skin rash. Less commonly, patients have symptoms of diabetes insipidus (DI), respiratory insufficiency, cytopenias, lymphadenopathy, liver dysfunction, or organomegaly.
In the past, three distinct clinical syndromes were described: eosinophilic granuloma, Hand-Schüller-Christian disease, and Letterer-Siwe disease. “Eosinophilic granuloma,” a term still commonly used by radiologists and orthopedists, refers to the presence of one or more lytic bone lesions. Hand-Schüller-Christian disease consists of the triad of bone defects, exophthalmos, and polyuria. Letterer-Siwe disease, a fulminant disorder of the reticuloendothelial system, is characterized by hepatosplenomegaly, lymphadenopathy, skin rash, bone lesions, anemia, and the tendency to bleed. This classification has important historic significance, but its applicability in the clinical setting is limited. Once it was recognized that the diverse manifestations of the disease share common histopathologic features, the three syndromes were unified under the term “histiocytosis X.” Categorizing patients based on the number and location of lesions and the presence or absence of organ involvement was shown to be useful in predicting prognosis and determining therapy. Single-system LCH (SS-LCH) disease—which usually affects bone, less frequently affects skin, and rarely affects the lymph nodes, lung, or central nervous system—accounts for approximately two thirds of pediatric LCH cases. Involvement of two or more organ systems, referred to as multisystem LCH (MS-LCH), accounts for the remaining one third of cases. In about half of MS-LCH cases, “risk” organs (i.e., the liver, spleen, and hematopoietic system) are affected. Thus far the presence of BRAF V600E does not correlate with disease extent or prognosis.
Pathology
A biopsy of lesional tissue is required to establish the diagnosis of LCH. The pathologic diagnosis is usually straightforward, but because the disease is rare, varied, and may mimic many other conditions, delay in diagnosis is common. LCH should be considered in any patient who presents with skeletal lesions, a persistent rash, chronically draining ears, central DI, unexplained lymphadenopathy, respiratory insufficiency with a reticulonodular pattern on a chest radiograph, hepatosplenomegaly, or cytopenias.
Once clinical suspicion is raised, the workup should proceed with a biopsy of the most accessible site of disease. Complete surgical excision of the lesion is generally unnecessary either for diagnosis or treatment. LCH lesions at all sites share common histopathologic features: accumulation of large neoplastic LCs with a moderate amount of dense pink cytoplasm and plump, distinctive “C-shaped,” “coffee bean,” or cleaved nuclei admixed with variable numbers of inflammatory cells, including T lymphocytes, macrophages, plasma cells, and eosinophils. The composition and architecture of the infiltrate characteristically differ according to the location. Osteolytic bone lesions often contain many nonneoplastic osteoclasts, as well as multinucleated tumor giant cells and large numbers of eosinophils, the latter speaking to the origin of the term “eosinophilic granuloma.” Mature, “burned out” bone lesions may be difficult to distinguish from chronic osteomyelitis, which is often in the clinical and radiographic differential diagnosis. LCH infiltration of the skin typically involves the superficial dermis, with tumor cells infiltrating the epidermis (so-called “epidermotropism” or “exocytosis”), thus altering epidermal barrier function and leading to superinfection and the characteristic appearance of some LCH rashes. In all cases, the neoplastic LCs express the LC markers CD1a and langerin, as well as S100 protein and fascin ( Fig. 64-1 ). Electron microscopy, which historically is used to demonstrate the presence of Birbeck granules indicative of the LC lineage, has little diagnostic utility at the present time. Cytogenetics or molecular studies are predominately needed to exclude other diseases with similar clinical presentations or histologies. Furthermore, no immunohistochemical or other laboratory methods are useful in distinguishing clinically indolent from clinically aggressive forms of the disease.
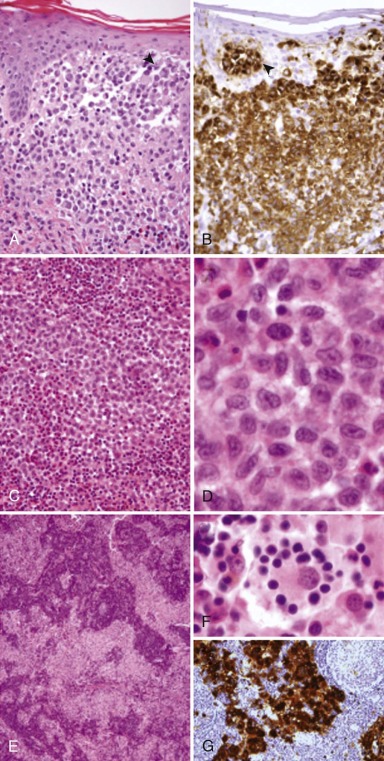
Diagnostic Evaluation
In suspected LCH cases, evaluation for disease extent is indicated, starting with a history and physical examination. A skeletal radiographic survey is recommended to comprehensively assess skeletal involvement. Abnormalities detected on plain radiographs may be followed by axial imaging to document the presence of a soft tissue mass. Nuclear imaging (i.e., a bone scan or fluorodeoxyglucose positron emission tomography [FDG-PET]) complements the skeletal survey. Compared with plain radiographs, nuclear imaging tests detect active lesions on the basis of increased local metabolic activity. Active bone lesions usually show increased radiotracer uptake, whereas inactive lesions may be undetectable or appear “photopenic.” FDG-PET appears to be the most sensitive imaging modality for the detection of lesions in persons with LCH, and it is particularly useful for following up on patients. Unlike a bone scan, PET is capable of detecting extraosseous lesions.
Complete blood cell counts and liver function studies should be performed, and liver and spleen size should be assessed by physical examination. Abdominal ultrasound is indicated when laboratory studies are abnormal or if hepatosplenomegaly is present or suspected. The erythrocyte sedimentation rate may be elevated, but it is not a sensitive or specific disease indicator. Measurement of specific gravity of an early morning urine sample is an easy and inexpensive screening test for DI. A formal water deprivation test may be necessary to establish the diagnosis of DI. Magnetic resonance imaging (MRI) of the head should be performed in children with cranial bone lesions and when DI is suspected or confirmed. A chest radiograph should be performed in all cases, and a computed tomography (CT) scan of the chest should be performed if the radiograph findings are abnormal or if the patient has respiratory signs or symptoms. Additional evaluations such as upper/lower endoscopy and biopsies of bone marrow, skin, lymph nodes, and the liver should be considered in the appropriate clinical context.
Management
The approach to management of LCH must account for the variability in its clinical behavior. On one hand, LCH is an uncontrolled accumulation of a clonal population of cells, with the capacity to behave extremely aggressively and the potential to involve multiple sites and organ systems. The process may be driven, in part, by oncogenic variants of BRAF . In addition, it may be effectively treated with cytotoxic chemotherapy and radiotherapy and, based on preliminary reports, BRAF antagonists. On the other hand, LCH is histologically benign, it sometimes resolves spontaneously, and it may respond to immunomodulatory or immunosuppressive agents. The historic uncertainty about the fundamental nature of LCH is reflected in the broad range of therapies that have been used to treat it.
Performing controlled clinical trials in persons with LCH has been challenging. As a consequence, the LCH literature is often descriptive, anecdotal, and retrospective. Since the 1980s, several collaborative groups have conducted prospective clinical trials that have led to standardization and improvements in treatment. An overall observation of these studies is that the treatment of LCH should match the clinical scenario. Patients with localized disease have an excellent prognosis with little or no intervention. For these children, the goal of therapy is to minimize symptoms and avoid long-term disability and potential late effects. In contrast, children with MS-LCH, especially those with involvement of the hematopoietic system or liver, are at significant risk for morbidity and mortality. For these children, aggressive treatment is justified.
Bone
Bone is the most commonly involved site, with bone involvement occurring in approximately 75% of children with LCH. Although any bone(s) may be affected, the bones of the skull, pelvis, and vertebral bodies are most commonly affected ( Fig. 64-2 ). Bone pain (which is often worse at night), swelling, and a limp are typical presenting symptoms. Sometimes a history of local trauma precedes or coincides with the onset of symptoms. The typical plain radiographic appearance is of a smooth-edged, “punched-out” hole in the bone. Lesions of the long bones typically involve the diaphysis, but metaphyseal and epiphyseal lesions also occur. Involvement of a vertebral body may manifest as flattening or loss of height (vertebra plana) or as a wedge deformity. Axial imaging (CT and MRI) may show an associated soft tissue mass ( Fig. 64-3 ). The radiographic differential diagnosis of osseous LCH lesions includes malignancy, especially Ewing sarcoma, leukemia or lymphoma, and osteomyelitis.
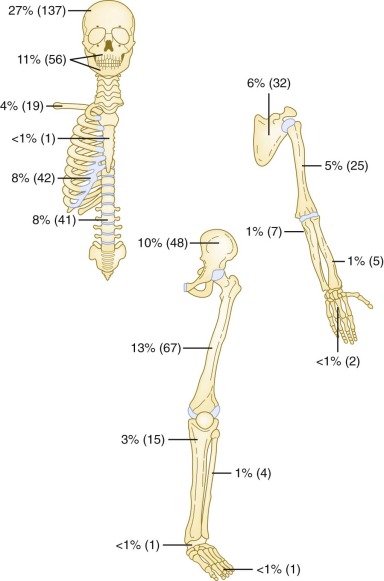
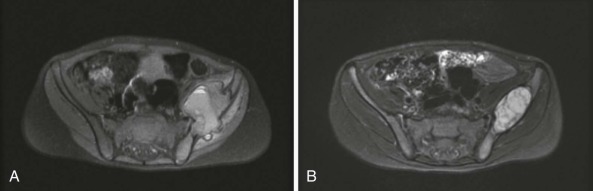
LCH of the bone may be unifocal (monostotic) or multifocal (polyostotic), and it may present in association with disease in other organ systems. Monostotic disease, which is the most common presentation of LCH (especially in older children), occurs in more than 50% of cases. Children with LCH that is limited to a single bone have an excellent prognosis regardless of the treatment administered. Although various local and systemic therapies appear to be efficacious, retrospective trials have failed to demonstrate a comparative advantage for any specific intervention or modality, including observation only, biopsy, curettage, simple excision, intralesional steroid instillation, local radiotherapy, or systemic therapy.
Often the biopsy procedure performed to establish the diagnosis of LCH of the bone provides therapeutic benefit. In many cases, adequate tissue for diagnosis may be obtained via percutaneous needle biopsy with ultrasound or CT guidance. Neurosurgeons tend to prefer open biopsy of orbital and skull base lesions and simple excision or curettage of calvarial lesions. Regardless of the surgical approach, most patients experience progressive healing after the procedure, even when complete excision of the lesion is not achieved. In cases in which LCH is strongly suspected or confirmed intraoperatively via frozen section, methylprednisolone can be instilled directly into the lesion. This intervention yields rapid relief of pain and may hasten resolution. Intralesional steroid injection is less commonly used in the management of cranial and vertebral lesions.
In the past, radiotherapy was routinely used to treat bone lesions. Even though a relatively low dose in the range of 600 to 1000 centigray is effective, the role of radiotherapy in pediatric LCH has waned because the long-term risk of radiotherapy, especially the risk of secondary malignant neoplasms, is rarely justified in treatment of a benign disease. In the setting of disseminated disease, local radiotherapy contributes little benefit.
Systemic therapy may be required for patients with monostotic LCH who experience unrelenting symptoms or are at significant risk for serious or permanent disability related to the location or extent of their lesion. This principle applies in patients with unifocal craniofacial bone involvement, including the orbit, temporal bone, mastoid, sphenoid, zygomatic, ethmoid, maxilla, paranasal sinuses, or cranial fossa (so-called “central nervous system [CNS]-risk” lesions), because these children are at increased risk for DI and other neurologic sequelae. Currently it is standard practice to administer chemotherapy in “CNS-risk” cases, even though it has not been definitively established that treatment decreases the risk of neurologic complications.
Systemic therapy is also recommended for children with polyostotic (i.e., multifocal bone) LCH. In these patients, an important goal of treatment is to mitigate the risk of future reactivation, because reactivation risk is approximately 40% for patients with multifocal bone disease versus 10% for patients with unifocal bone disease. Children with SS-LCH of the bone who require systemic therapy—that is, those with CNS-risk lesions or multifocal bone involvement—are typically treated with the combination of vinblastine and prednisone (discussed later).
Skin
The skin is the second most commonly involved system in persons with LCH, with skin involvement occurring in about one third of patients. Skin rashes may be extremely variable, and misdiagnosis is common. Cutaneous LCH may present as single or multiple nodules, vesicles, or scaly, seborrhea-like patches or plaques. Although the rash may be located anywhere on the body, it is typically most intense in the flexural areas, such as the neck, axillary, and inguinal folds. Involvement of the scalp, the ear canals, and the posterior auricular areas is also characteristic. Ear canal involvement may lead to chronic ear drainage that may be malodorous. Perineal and perianal rashes may be severe, persistent, and refractory to topical preparations.
Cutaneous involvement is more common in infants than in older children. Some infants with isolated cutaneous LCH prove to have a benign, self-healing disorder known as Hashimoto-Pritzker disease. However, in infants who initially present with apparently isolated cutaneous LCH, MS-LCH may proceed to develop, and therefore self-healing cutaneous LCH is a diagnosis that can be made with accuracy only in retrospect. The differential diagnosis of cutaneous LCH may include superficial candidiasis, seborrheic dermatitis (“cradle cap”), eczema, contact dermatitis, and viral exanthem. A skin biopsy is required to establish the diagnosis.
When treatment for cutaneous LCH is required, topical medications such as corticosteroid or nitrogen mustard may suffice. Occasionally, systemic therapy is necessary to control extensive or severe skin involvement, especially when it is associated with pain, disfigurement, or infection. Alternatives such as psoralen ultraviolet A or interferon have been used with success in some patients who have resistant cutaneous involvement.
Central Nervous System
The brain and skull can be affected by LCH in a variety of ways, including extension of extraaxial bone-based lesions, hypothalamic-pituitary disease, intraparenchymal mass lesions, and later onset neurodegeneration.
Hypothalamic-pituitary disease is the most common and best-characterized CNS manifestation of LCH. Infiltration of the pituitary stalk by lesional cells leads to deficiency of antidiuretic hormone and clinical DI. Anterior pituitary deficiencies or panhypopituitarism may develop in some cases. The diagnosis of DI may precede, coincide with, or occur years after the diagnosis of LCH. Estimates of the frequency of DI in persons with LCH vary considerably because of variability in study populations. One large retrospective review reported a 12% incidence of DI, with 6% having DI present at the time of LCH diagnosis. In this analysis, the risk of DI correlated significantly with the location and extent of LCH involvement. Children who have craniofacial lesions (CNS risk) or MS-LCH are at significantly higher risk for DI.
Among children with a new presentation of DI, LCH is the cause in a significant proportion (approximately 15%) of cases. Absence of posterior pituitary hyperintensity (the pituitary “bright spot”) or thickening of the pituitary infundibulum may be noted on brain imaging; however, these radiographic findings are neither sensitive nor specific for LCH. Their differential diagnosis includes brain tumors, especially germinoma, and inflammatory conditions such as hypophysitis. Investigation for other sites of LCH is worthwhile because detection and biopsy of extracranial lesions may allow histopathologic confirmation of the diagnosis without performing a pituitary stalk biopsy.
Screening for DI in children who have LCH with use of a careful history is essential, and referral to an endocrinologist should be considered if symptoms of polyuria, polydipsia, nocturia, or dehydration are elicited. Urinary specific gravity greater than 1.015 weighs against a diagnosis of DI, and this reading can often be easily determined by analysis of a first-morning voided urine specimen. In cases in which the history or laboratory studies raise suspicion, a formal water deprivation test should be performed.
In general, DI is irreversible once it appears in patients with LCH. However, improvement has been reported in “early” DI cases when patients were treated promptly with chemotherapy. At one time, emergent radiotherapy was recommended in patients with new onset DI. This recommendation has fallen out of practice because it is rarely, if ever, effective and is associated with potential long-term consequences. Chemotherapy has no role in patients with established, long-standing DI who have no evidence of active LCH within or outside of the CNS.
Parenchymal CNS mass lesions in persons with LCH are composed of granulomas of CD1a+ cells admixed with other inflammatory cells. These lesions can occur in isolation or in association with disease in other organ systems. Depending on the extent and location, headaches, seizures, and focal neurologic symptoms may result. CNS mass lesions are managed with chemotherapy, surgery, or radiotherapy.
Progressive neurodegeneration is an uncommon and poorly understood late event that arises years after LCH diagnosis. Radiographically, it is characterized by signal abnormality confined to the brain stem and cerebellum on MRI. Posterior fossa symptoms or neurocognitive dysfunction may be clinically apparent. Biopsy results show gliosis, neuronal cell loss, and lymphocytic infiltration without active CD1a+ cell infiltration. The timing and course of this manifestation of LCH is quite variable, but it may be severe and progressive. No established treatment exists for neurodegeneration in patients with LCH. Retinoic acid or low-intensity chemotherapy with or without immunoglobulin may stabilize the disease or slow the rate of neurologic decline.
Other Sites
Lymph node involvement is occasionally present in the setting of MS-LCH, and sometimes it represents a single site of disease. The pathologic pattern of lymph node involvement is characteristically interfollicular and subtle.
The gastrointestinal tract is another uncommon site of disease in LCH, but its incidence may be underappreciated. When it does occur, it is usually in the context of multisystem involvement. Clinical signs include diarrhea, bloody stools, failure to thrive, and hypoalbuminemia. Gastrointestinal biopsies typically demonstrate a sparse infiltrate of LCs in the lamina propria.
Pulmonary involvement was historically thought to be an adverse prognostic feature, warranting its classification as a “risk” organ in early Histiocyte Society clinical trials. Involvement of the lung may be asymptomatic in up to half of affected children, with diffuse micronodular interstitial disease or cyst formation. Overt respiratory symptoms including tachypnea, chronic cough, dyspnea, and pneumothorax are seen in some patients. In children, pulmonary involvement occurs in the context of MS-LCH and is often accompanied by hematologic or hepatic dysfunction. Recent retrospective studies have shown that in the absence of other risk organ involvement, pulmonary involvement is not an independent negative prognostic indicator. Interestingly, adults with LCH often present with disease limited to the lung, and their illness is closely linked to cigarette smoking. Cessation of smoking leads to resolution in the majority of patients. Although traditionally described as a polyclonal disease, as many as 30% of cases may be clonal, and a significant proportion harbor BRAF V600E.
Risk Organs
Although the behavior of LCH is unpredictable, it is known that children with disease involving the hematopoietic system or liver are at highest risk for severe illness and death. Conventionally, involvement of “risk” organs is defined by the presence of organ dysfunction, such as cytopenias, hyperbilirubinemia, and impaired hepatic synthetic function. Risk organ involvement is typically a component of MS-LCH and is often associated with overt constitutional symptoms. Involvement of one or more risk organs occurs in approximately half of children with MS-LCH. Risk organ involvement is usually present at diagnosis, but it can develop later. It is clearly associated with young age, because more than half of children younger than 2 years manifest risk organ involvement. The association between young age and aggressive disease accounts for the perception that young age is an adverse prognostic feature.
The presence of peripheral blood cytopenias in a patient with LCH is referred to as hematologic dysfunction and is taken to be synonymous with bone marrow involvement. Interestingly, however, evidence of bone marrow infiltration by morphologically apparent LCs is not typically seen. Most often the marrow contains a lymphohistiocytic infiltrate with hemophagocytosis that can be highly reminiscent of hemophagocytic syndromes (which will be discussed later), further illustrating that the neoplastic LCs retain some of the immunomodulatory function of their normal counterparts. Splenomegaly is commonly seen in association with hematologic dysfunction as a consequence of organ infiltration, extramedullary hematopoiesis, and hemophagocytosis. Hypersplenism may contribute to cytopenias in these patients.
Hepatic dysfunction, like hematologic dysfunction, is associated with serious illness and a high risk of morbidity and mortality. Hepatic dysfunction is associated with any of the following findings: hepatomegaly, ascites, hyperbilirubinemia, increased serum concentrations of hepatic transaminases (especially γ-glutamyl transferase) and impaired hepatic synthetic function (hypoalbuminemia and hypoproteinemia). The pathogenesis of liver involvement is heterogeneous. In some cases hepatomegaly or transaminitis is a consequence of macrophage activation in a sinusoidal pattern in the liver and is associated with multisystem disease. In other cases, a cholestatic laboratory picture predominates, presumably resulting from the infiltration of hepatic bile ducts by CD1a+ LCs, even if these cells are not demonstrable upon a percutaneous liver biopsy. In these patients, progressive hepatic dysfunction may lead over time to irreversible hepatic cirrhosis that requires liver transplantation, even after resolution of active LCH.
Treatment of Multisystem LCH
Systemic therapy is indicated for all patients with MS-LCH and for some patients with SS- LCH, as previously described. The optimal therapy for this diverse group of patients is still not established. Over the years, a wide variety of immunosuppressive and cytotoxic drugs have demonstrated activity. Encouraging results were first published in the early 1960s using corticosteroids and vinblastine. Later, alkylating agents and antimetabolites were used. In the 1980s, the epipodophyllotoxin etoposide was shown to have particular cytotoxicity for cells of the monocyte/macrophage lineage. This agent showed promising results in children with refractory LCH and was subsequently studied in newly diagnosed patients. More recently, the nucleoside analogs 2-chlorodeoxyadenosine (2-CdA) and clofarabine have shown promise in children with refractory LCH and in adults. Regardless of the treatment regimen, survival among low-risk patients is excellent. However, a substantial proportion of high-risk patients still experience a progressive and fatal course despite therapy.
The Deutsche Arbeitsgemeinschaft fur Leukamieforschung und Behandlung im Kindersalter (German Working Group for Research and Treatment of Leukemia in Childhood) performed two consecutive multicenter studies (known as DAL-HX 83 and DAL-HX 90) in which children with multifocal bone or MS-LCH were treated with a nonrandomized, risk-adapted, prospective, multidrug regimen. After initial therapy with prednisolone, vinblastine, and etoposide, children who had multisystem disease went on to receive 1 year of continuation therapy with prednisolone, vinblastine, etoposide, and mercaptopurine. In the DAL-HX 83 study, patients with organ dysfunction also received methotrexate. In these trials, approximately 67% of the patients who had organ dysfunction responded to therapy, with 42% of these patients experiencing subsequent disease reactivation. In contrast, the patients with multisystem disease who did not have organ dysfunction and the patients with multifocal bone disease had a roughly 90% response rate and a 20% reactivation rate. Unfortunately, death due to refractory or progressive LCH occurred in about 20% of subjects in the DAL-HX studies. Mortality was restricted to children with disseminated disease, organ dysfunction, and an unfavorable response to initial therapy.
In 1991, The Histiocyte Society initiated LCH I, the first international, randomized clinical trial in persons with LCH. This study compared the efficacy of 6 months of vinblastine (arm A) versus etoposide (arm B) in children with multisystem disease. The results showed that the two treatment arms, arms A and B, were equivalent in all respects, including response at week 6 (57% and 49%), toxicity (47% and 58%), probability of survival (76% and 83%), probability of disease reactivation (61% and 55%), and probability of developing permanent sequelae (39% and 51%), including DI (22% and 23%). Of the 29 children in the study who died as a result of their disease, all had risk organ involvement. The probability of survival among the children older than 2 years who did not have risk organ involvement was 100%. Secondary acute myeloid leukemia developed in one patient in arm B. In light of the fact that etoposide was not superior to vinblastine in any measure, the study suggested that the added risk associated with it may not be justified.
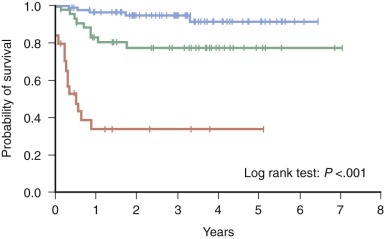
Importantly, analysis of the LCH I study revealed that early response to therapy is an extremely powerful prognostic indicator in persons with MS-LCH. Children younger than 2 years who had MS-LCH and failed to respond to 6 weeks of initial therapy had a dismal 17% survival rate compared with an 88% survival rate in children who responded favorably ( Fig. 64-4 ). The prognostic value of early response was confirmed in a retrospective analysis of the DAL-HX 83 and DAL-HX 90 studies.
The Histiocyte Society’s successor study, LCH II, intensified treatment for all patients and further explored the role of etoposide in persons with MS-LCH. All children received 6 weeks of initial therapy that consisted of continuous oral prednisone and weekly administration of vinblastine. Low-risk children who were risk organ negative received continuation therapy with pulse prednisone and vinblastine every 3 weeks for a total of 6 months. High-risk children who were risk organ positive also received mercaptopurine and were randomly assigned to either receive etoposide (arm B) or not receive etoposide (arm A) during both the initial and continuation phases. LCH II did not demonstrate a statistically significant benefit of the addition of etoposide in patients with MS-LCH; response at week 6 (63% vs. 71%), survival (74% vs. 79%), disease reactivation (46% in both arms) and permanent consequences (43% vs. 37%) were all similar in arms A and B, respectively. However, in comparison with the less-intensive therapy studied in the LCH I trial, the more intensive LCH II regimen did show a somewhat higher rapid response rate and lower mortality in children with risk organ involvement.
LCH III opened for enrollment in 2001. For the highest risk group of subjects in this study—that is, the children with MS-LCH who had involvement of risk organs—methotrexate was added to the backbone of prednisone, vinblastine, and mercaptopurine in a randomized fashion. The total duration of therapy was lengthened to 12 months for all high-risk patients. Children without risk organ involvement were randomly assigned to receive vinblastine and prednisone for 6 versus 12 months. The study failed to demonstrate a benefit as a result of the addition of methotrexate to the standard backbone in multisystem high-risk patients. However, prolongation of therapy from 6 to 12 months in multisystem low-risk patients is associated with a significantly lower reactivation rate (a 3-year cumulative incidence of reactivation of 50% vs. 35%).
Refractory Langerhans Cell Histiocytosis
Children with MS-LCH whose disease does not respond promptly to vinblastine and prednisone have an unfavorable prognosis, with survival rates in the range of 10% to 34%. Unfortunately, LCH that is resistant to vinblastine and corticosteroid is often unresponsive to other conventional agents. 2-CdA, a nucleoside analog, has shown significant promise as salvage therapy in children with reactivated or refractory disease. Dramatic responses to 2-CdA combined with cytarabine have been observed in children with refractory LCH who have hematologic dysfunction. Also, the nucleoside analog clofarabine has demonstrated activity in a smaller number of reported cases of refractory MS-LCH.
A small but growing number of children with refractory MS-LCH have been successfully treated with allogeneic hematopoietic cell transplantation (HCT). Not surprisingly, toxicity is a significant obstacle in these very ill patients. Autologous transplantation has not resulted in durable responses. Transplant regimens that incorporate reduced intensity conditioning are especially appealing in persons with refractory LCH and appear to be feasible, with lower transplant-related morbidity and mortality.
Reactivation
Some children experience LCH reactivation after initial improvement or resolution of their disease. Most reactivations occur within 1 year of diagnosis, and almost all develop within 2 years. Patients with MS-LCH are clearly at higher risk of reactivation than are patients with SS-LCH. Patients occasionally experience a chronic, relapsing, and remitting course with repeated reactivations. Aggressive radiographic surveillance for reactivation is generally discouraged, because the site of reactivation is unpredictable and the risk of imaging may outweigh the benefit of early diagnosis. However, monitoring physical examination and growth and development is advised, along with prompt evaluation of any new signs or symptoms. Elevation of the erythrocyte sedimentation rate and the platelet count can be a tip-off to disease reactivation.
Bone is the most frequent site of LCH reactivation. Fortunately, reactivation in risk organs is distinctly uncommon in children who did not have risk organ involvement at presentation. Localized reactivations, such as a single bony site or recurrent rash, may be effectively managed with local therapy alone (i.e., surgery or intralesional or topical administration of a corticosteroid). However, systemic therapy is often necessary in persons with reactivated disease to control symptoms, prevent permanent sequelae, and decrease the severity and frequency of subsequent reactivations. In contrast to recurrent malignancies in which acquired drug resistance limits the efficacy of cytotoxic drugs after previous exposure, in reactivated cases of LCH, medications used successfully in the setting of newly diagnosed disease often remain effective in the treatment of reactivation. When cytotoxic therapies such as corticosteroids, vinca alkaloids, antimetabolites, or nucleoside analogues fail to provide durable disease control or when adverse effects are limiting, more intensive and less conventional options are worth exploring. For these relatively rare cases, therapy must be individualized, keeping in mind the specific site(s), symptoms, and risk of the reactivation, as well as the specific adverse effects of therapy.
Alternatives to conventional therapy that have been used in bone reactivation include nonsteroidal antiinflammatory agents (e.g., indomethacin) and bisphosphonates (e.g., pamidronate). These agents may act to ameliorate pain but may not directly inhibit disease activity. Thalidomide has shown some efficacy in patients with low-risk LCH that is refractory to conventional therapy. The presumed mechanism of action is by inhibition of TNF or other cytokines. Many children who undergo repeated reactivations experience permanent disabilities related to their LCH. Nonetheless, their overall likelihood of survival approaches 100%.
Long-Term Consequences
Roughly half of survivors of LCH experience irreversible long-term consequences of their illness or, to a lesser extent, of the therapy they received. The incidence and pattern of sequelae strongly correlate with the initial extent and pattern of disease involvement; patients who experience multisystem disease and multiple reactivations are at highest risk. A considerable fraction of irreversible complications observed in survivors of LCH were present at the time of diagnosis, and thus not all long-term complications are preventable.
Orthopedic complications are the most frequently observed permanent sequelae in survivors of SS-LCH. Examples include spinal deformity leading to scoliosis or kyphosis, dental loss due to involvement of the jaw, and facial asymmetry or proptosis due to involvement of orbital or facial bones. Although conductive hearing loss attributed to chronic inflammation of the middle and external ear is reversible, sensorineural hearing loss due to involvement of osseous structures is usually permanent when it occurs. Although permanent orthopedic and cosmetic consequences are relatively frequent, they do not necessarily adversely affect the quality of survival.
DI is an irreversible condition in almost all cases in which it occurs, and a lifelong need for desmopressin is to be expected. Similarly, other endocrinopathies such as growth hormone deficiency and panhypopituitarism are not responsive to LCH-directed therapy but are managed with hormone supplementation. Awareness and close attention to growth and development are very important in the follow-up care of these patients.
Rarely, survivors of LCH experience progressive end organ damage in the absence of demonstrable activity of LCs. Progressive hepatic cirrhosis with liver failure requiring liver transplantation is a rare but well-recognized late event. A somewhat analogous process may occur in the lungs, leading to pulmonary fibrosis, cystic changes, or spontaneous pneumothorax. Adult survivors of pediatric LCH should be counseled to avoid exposure to cigarette smoke because of the potentially increased risk of developing pulmonary abnormalities. Neurodegeneration, which was previously described, is the most dreaded late event in survivors of LCH.
Overview of Therapeutic Options
Approach to SS-LCH
Children with localized SS-LCH have an excellent prognosis. Most children with localized disease may be safely observed after a biopsy or resection is performed, without the need for additional intervention. Local therapy may be added to accelerate resolution of symptoms. In patients with craniofacial bone or multifocal bone disease or isolated CNS involvement, systemic therapy is indicated to control symptoms and to prevent reactivations and long-term (especially orthopedic and neuroendocrine) sequelae.
Approach to Low-Risk MS-LCH
Children with MS-LCH who do not have involvement of risk organs have an excellent prognosis. Because their course is generally favorable, maximizing response while minimizing short- and long-term toxicities is the primary objective. When possible, enrollment in a clinical trial is encouraged. The current standard of care in the United States is the combination of vinblastine and prednisone for a total duration of 12 months.
Approach to High-Risk MS-LCH
Thus far, up-front intensification of therapy has not been shown to improve outcome in children with MS-LCH who have involvement of risk organs. Children with high-risk features who respond favorably to standard chemotherapy have a good prognosis, but children who do not respond rapidly to standard treatment have a poor prognosis. For this group of patients, intensive chemotherapy, investigational agents, or HCT should be considered.
The role of BRAF inhibitors in patients whose LCH cells harbor oncogenic BRAF variants remains to be established. The increased risk of squamous cell carcinomas of the skin associated with the currently available inhibitors will likely restrict their use to patients with a poor prognosis in whom the benefits may outweigh the risks.
Non–Langerhans Cell Histiocytoses
In addition to LCH, a spectrum of other even rarer histiocytic lesions exists that affect both children and adults. In general they include a large number of clinically benign, not infrequently progressive dermatologic disorders that are pathologically characterized by proliferations of Langerhans-like cells, often with distinctive cytosolic features (see Fig. 64-1 ). Other rare non-LCHs, most notably Erdheim-Chester disease and the “true” malignancies such as Langerhans cell sarcoma and histiocytic sarcoma, have high mortality rates. With the exception of JXG and sinus histiocytosis with massive lymphadenopathy (SHML), these disorders will not be discussed further because of their rarity, and the reader is referred to several authoritative reviews on these diseases.
Juvenile Xanthogranuloma
JXG is a generally benign tumoral proliferation of histiocytes with a CD1a−, S100−, Langerin−, CD68+, CD163+, fascin+ immunophenotype that, like LCH, is believed to be derived from dermal interstitial DCs. Lesions characteristically present as reddish to yellowish-brown cutaneous nodules in the head and neck region of children younger than 1 year of age; the median age of onset is 5 months, and tumors may be congenital. Multifocality is not uncommon, particularly in male infants. An association exists with neurofibromatosis 1, Noonan syndrome, and juvenile myelomonocytic leukemia. The natural history of most lesions is spontaneous regression, but they are often diagnosed by excisional biopsy, leaving little or no residual tumor. Extranodal involvement of the deep soft tissues, liver, spleen, conjunctivae, and the CNS—so-called systemic JXG—with or without cutaneous involvement occurs in nearly 5% of cases. Although systemic lesions also typically regress, local disease, as in the eye and CNS, may lead to significant morbidity. Patients with systemic disease involving the CNS or liver have occasionally died of their disease. In cases requiring treatment, single agents or multiagent regimens similar to those used for persons with LCH have been employed with success.
Sinus Histiocytosis with Massive Lymphadenopathy
SHML, also known by the eponym Rosai-Dorfman disease, is an enigmatic histiocytic disorder associated with a proliferation of CD1a−, Langerin−, S100+, CD68+, CD163+, fascin+ histiocytes with abundant foamy cytoplasm. Most characteristically, a subset of lesional histiocytes contains a large number of lymphocytes and other cells, but unlike in the hemophagocytic syndromes (discussed later), these cells are not contained within phagolysosomes. Rather, they are “just passing through” in invaginations of the cell membrane, in a process termed emperipolesis (see Fig. 64-1 ). In lymph nodes, these histiocytes preferentially involve the nodal sinuses and secondarily the interfollicular areas, leading to remarkably huge lymphadenopathy, which gives the disease its name.
Because the SHML literature is largely retrospective, particularly since 1990 when a patient registry was last updated, the reported patterns of clinical presentation and natural history of the disease are undoubtedly biased. Nevertheless, the prototypical clinical presentation is massive, painless cervical lymphadenopathy. In addition, up to 40% of patients present with extranodal disease, most commonly in the skin, upper respiratory tract, and bone. The orbit and meninges are also not uncommon extranodal sites. Although extensive nodal and extranodal (especially kidney, lower airway, and liver) disease and coexisting immunologic disorders tend to correlate with an adverse prognosis, it is apparent that many if not most cases remit spontaneously without therapy. Constitutional symptoms including fever associated with neutrophilia, increased erythrocyte sedimentation rate, and polyclonal hypergammaglobulinemia that are often associated with SHML usually respond to systemic corticosteroids. In cases in which disease has compromised organ function, requiring therapy, no treatment modality is clearly preferred; surgical debulking and radiation therapy generally result in some amelioration, but the response to systemic chemotherapy—often a combination of alkylators, vinca alkaloids, and steroids—is inconsistent.
Monocytes and Macrophages
Resident tissue macrophages play essential roles in host defense, especially when they populate tissues that face the external environment (e.g., alveolar macrophages in the lung). However, macrophages also contribute to tissue homeostasis through their repair and clearance activities. Functional and morphologic heterogeneity among macrophages has been long recognized, and identification of specific subsets has been aided by antibody-based identification of characteristic cell surface markers. Some of these subsets may be involved in the histiocytoses of macrophage origin.
Among the macrophages active in host defense and inflammation, alveolar and gut macrophages are best understood. These cells express pattern recognition receptors and scavenger receptors that enhance recognition of microbial products, are highly phagocytic, and are efficient antigen presenters. However, even within this category some degree of heterogeneity exists because gut macrophages tend to secrete lower amounts of proinflammatory cytokines. Kupffer cells are derived from macrophages and may also play a primarily defensive role in the host. In contrast, macrophages involved in homeostasis include osteoclasts and microglia.
Macrophages in secondary lymphoid organs show additional degrees of heterogeneity, with specific cell types occupying characteristic anatomic niches. In the spleen, red pulp macrophages, which express high levels of the murine surface marker F4/80, are distinguishable from tingible body macrophages in the white pulp. Also present in the white pulp are metallophilic macrophages, which align adjacent to the marginal sinus. Within the marginal zone itself are functionally distinct macrophages that express pattern recognition and scavenger receptors they may use to clear pathogens from the bloodstream. These macrophages express much lower levels of the F4/80 marker than do red pulp macrophages. Similar subanatomic heterogeneity is observed in lymph nodes: macrophages in cortical regions express low levels of F4/80, whereas macrophages in the medullary sinuses, paracortex, and subcapsular sinuses express high levels of F4/80.
In the absence of specific stimuli, tissue macrophages remain in a low-functioning, resting state. Activation induces functions that are characteristic of their host protection or homeostasis roles. Based on in vitro analyses, four distinct pathways of macrophage activation have been described. “Classical” activation via interferon-γ or lipopolysaccharide induces microbicidal activities and upregulation of major histocompatibility complex class II expression. “Alternative” activation via IL-4 or IL-13 induces expression of genes that are involved in tissue repair or suppression of inflammation. “Innate” activation via toll-like receptor ligands also induces microbicidal activities. Finally, “deactivation” via IL-10 or transforming growth factor–β stimulation reduces class II expression and increases secretion of antiinflammatory cytokines. As clearly defined as these pathways are in vitro, little evidence exists as yet that they are relevant in vivo. However, secretion of cytokines by activated macrophage-derived cells may be a major pathophysiologic factor in histiocytoses. The first and third mechanisms lead to the production of what are sometimes called “M1” macrophages, whereas the second and fourth mechanisms produce “M2” macrophages.
Origins of Macrophages
For the most part, steady-state renewal of tissue macrophages appears to be accomplished by local proliferation of similar cell types. However, under conditions of rapid turnover such as inflammatory states, macrophages are replaced by bone marrow precursors—that is, circulating monocytes. Recent findings have revealed substantial heterogeneity among circulating monocytes and the roles played by specific subsets in macrophage development.
Broadly defined, circulating monocytes can be separated into “classical” or inflammatory cells versus “resident” cells, which have a phenotype that closely resembles tissue macrophages. Cell surface markers can distinguish these subsets: inflammatory monocytes are CD14hi CD16− in humans and Ly6C+ in mice; resident monocytes are CD14+ CD16+ in humans and Ly6C− in mice. Experiments in mice have tracked the fate of some of these cells. For example, bone marrow–derived Ly6C+ cells, which express high levels of the chemokine receptor CCR2 and low levels of the chemokine receptor CX3CR1, enter the circulation. Ordinarily, these cells are very short-lived, but in inflammatory states, they are attracted to affected sites where they can differentiate into macrophages or DCs. Meanwhile, the precise process whereby circulating monocytes replenish resident macrophages (which are CCR2−, CX3CR1hi, Ly6C−) is unknown. Nonetheless, some of the signals that dictate tissue-specific macrophage differentiation are understood. For example, circulating mononuclear precursors that localize to bone will become osteoclasts under the influence of m-CSF and receptor activator of nuclear factor kappa-B ligand, which may have pathophysiologic relevance in histiocytic disorders that attack bone.
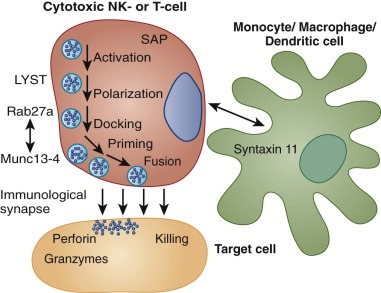
Hemophagocytic Lymphohistiocytosis
HLH is not a single disease entity but rather a syndrome consisting of clinical signs and laboratory abnormalities that result from the uncontrolled proliferation and activation of cells of the monocyte/macrophage lineage. Normally, monocytes and macrophages are responsible for phagocytosis of antigen and activation of other immune cells through the production of cytokines and chemokines. By way of their interactions with natural killer (NK) cells, macrophages are an important feature of the innate immune response. Additionally, they participate in adaptive immunity via their complex interactions with T cells. When pathogens, especially viral pathogens, are not rapidly eliminated by the innate immune response, the chemokines and cytokines produced by activated macrophages stimulate and perpetuate T-lymphocyte activation. Activated macrophages are themselves regulated by stimulatory and inhibitory factors that they and other immune cells produce. When this complex process escapes control, extremely potent and destructive inflammatory forces are unleashed. Uncontrolled macrophage activity is the hallmark of HLH. Although the pathogenetic mechanisms underlying the hyperinflammatory state in HLH are heterogeneous, the clinical consequences are shared.
The earliest published reports of hemophagocytic syndrome by Farquhar described four siblings who were afflicted in early infancy with fever, irritability, hepatosplenomegaly, and pancytopenia. Autopsy demonstrated histiocytic proliferation throughout the reticuloendothelial system with prominent hemophagocytosis. The disorder was dubbed “familial haemophagocytic reticulosis” and was recognized as distinct from Letterer-Siwe disease (i.e., the disseminated form of LCH). An autosomal-recessive pattern of inheritance was proposed. Subsequently, sporadic cases with similar clinical features were seen in association with viral infections, bacterial infections, and a host of other infections. Concomitantly, it was observed that certain systemic illnesses, notably rheumatologic conditions and malignancies, occasionally led to the development of clinical features of hemophagocytic syndrome. In 1987, the Histiocyte Society adopted the unifying term hemophagocytic lymphohistiocytosis and defined a set of diagnostic criteria to assist clinicians and researchers. The criteria have subsequently been refined to account for advances in our understanding of the syndrome ( Box 64-2 ).
Diagnostic Guidelines (1994) *
* All criteria required. If hemophagocytic activity is not proven at presentation, further search is encouraged.
Clinical Criteria
- •
Fever
- •
Splenomegaly
Laboratory Criteria
- •
Cytopenias (hemoglobin <9 g/L; platelets <100 × 109/L; neutrophils <1.0 × 109/L)
- •
Hypertriglyceridemia and/or hypofibrinogenemia (fasting triglycerides ≥3 SD, fibrinogen ≤3 SD of normal for age)
Histopathologic Criteria
- •
Hemophagocytosis in marrow, spleen, or lymph nodes
- •
No evidence of malignancy
Revised Diagnostic Guidelines (2004)
Molecular diagnosis consistent with hemophagocytic lymphohistiocytosis OR five of the following eight clinical, laboratory, and histopathologic criteria
Clinical Criteria
- •
Fever
- •
Splenomegaly
Laboratory Criteria
- •
Cytopenias (hemoglobin <9 g/L; platelets <100 × 109/L; neutrophils <1.0 × 109/L)
- •
Hypertriglyceridemia or hypofibrinogenemia (fasting triglycerides ≥3 standard deviations, fibrinogen ≤3 standard deviations of normal for age)
- •
Hyperferritinemia (>500 µg/L)
- •
Elevated CD25 (≥2400 U/L)
- •
Low/absent natural killer function
Histopathologic Criteria
- •
Hemophagocytosis in marrow, spleen, or lymph nodes with no evidence of malignancy
Incidence
HLH is broadly categorized into two forms: familial (FHL) and secondary (sHLH). Undoubtedly the delineation is an oversimplification, because significant clinical and perhaps even genetic overlap exists between the two forms. Importantly, FHL and sHLH may be indistinguishable at presentation.
In persons with FHL, an ordinary immune stimulus, such as a common viral exposure, triggers an unrestrained hyperinflammatory state as a result of the existence of an inherited, intrinsic defect in the patient’s immunologic effector function. FHL is genetically heterogeneous, resulting from mutations that disturb the function of one or more of several proteins that participate in lymphocyte cytotoxicity. Approximately 1 in 50,000 live births are estimated to be affected by FHL. A slight preponderance of males may be attributable to the occurrence of FHL in association with X-linked lymphoproliferative disorders.
In contrast, sHLH develops as a consequence of an intense immunologic stimulus in the form of an infection, malignancy, or inflammatory process in a person without an inherited immune defect. sHLH is more prevalent than FHL, but its true incidence in children and adults is not defined.
Clinical Features
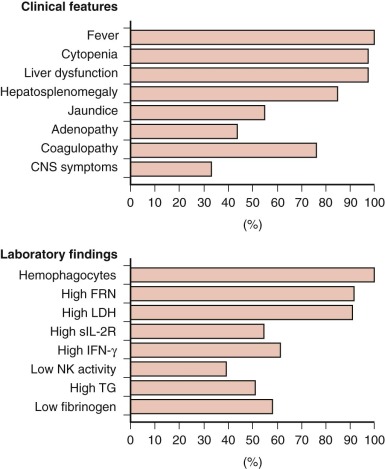
The cardinal clinical features of HLH are persistent high fever, cytopenias, and splenomegaly, with or without hepatomegaly. Unquestionably, this constellation of signs and symptoms is not rare or specific. Furthermore, establishing the diagnosis is made more difficult by the fact that all clinical features of the disorder may not be initially or simultaneously present ( Fig. 64-5 ).
Persistent and prolonged fever is essentially universal and is usually the first sign of illness. Signs of a routine childhood upper respiratory or gastrointestinal illness often accompany the fever. However, instead of resolving in the typical time course, constitutional symptoms generally progress in children who have HLH. At presentation, affected patients are usually acutely ill and require urgent evaluation and intervention.
Cytopenias that affect at least two cell lines are a key feature of HLH. Thrombocytopenia is almost always present. The platelet count may be normal or mildly depressed initially, but it often falls precipitously as the disease progresses. Normocytic anemia with reticulocytopenia is also common. In cytopenic patients, less than the expected increment may be seen in response to transfusions because of shortened survival of transfused blood cells. Leukopenia and neutropenia are more variably present.
Abdominal distention and hepatosplenomegaly are usually present and may be accompanied by jaundice and ascites. Biochemical evidence of liver dysfunction is typical, although it is not a universal feature of HLH. Hypertriglyceridemia, increased transaminases, or hyperbilirubinemia may be present and may in a very young child raise the possibility of primary liver disease or a metabolic disorder. Impaired hepatic synthetic function and clotting factor consumption leads to hypofibrinogenemia and prolongation of the prothrombin and partial thromboplastin times. When coagulopathy is combined with thrombocytopenia, the risk of bleeding escalates. Spontaneous hemorrhage, especially intracranially, may lead to acute decompensation and may raise an erroneous suspicion of child abuse.
Typically, the serum ferritin level is markedly elevated in patients who have active HLH. A serum ferritin level exceeding 10,000 µg/L is a highly sensitive and specific marker that supports the diagnosis. However, although most patients with HLH have an increased ferritin level, not all patients demonstrate extreme hyperferritinemia. Furthermore, mild to moderate elevation in the serum ferritin level may be seen in disorders other than HLH, such as inflammatory conditions, infections, liver disease, hemochromatosis, and metabolic disorders. The precise mechanism of ferritin elevation in persons with HLH is unknown, but it is probably related both to increased synthesis of ferritin mediated by high levels of cytokines and increased release of ferritin due to liver injury and red blood cell turnover.
In keeping with a state of generalized, uncontrolled inflammation, the levels of various cytokines are increased in patients with active HLH, including IL-2, IL-6, IL-8, IL-10, interferon, and TNF. Cytokine levels are not routinely measured, but the soluble IL-2 receptor (also known as CD25) is available as a clinical laboratory test. CD25 elevation is a sensitive and reliable marker of HLH activity, and it has been incorporated into the diagnostic criteria for HLH. CD25 levels are also elevated in other disorders, especially lymphoid malignancies, but typically to a lesser degree.
The term hemophagocytosis describes the pathologic finding of activated, histologically benign macrophages engulfing erythrocytes, leukocytes, platelets, and their precursor cells ( Fig. 64-6 ). This process is often assessed in the bone marrow, but it occurs throughout the reticuloendothelial system (i.e., in the liver, spleen, and lymph nodes) and sometimes in the CNS. Destruction of blood cells and their precursors leads to peripheral blood cytopenia(s), often in association with bone marrow hypercellularity. Analysis of the bone marrow is always recommended when a diagnosis of HLH is suspected, both to demonstrate hemophagocytosis and to rule out the presence of leukemia/lymphoma. Although the finding of hemophagocytosis in the bone marrow supports the diagnosis of HLH in the proper clinical context, it is not essential for the diagnosis or pathognomonic. In a series of 122 children with HLH from the Histiocyte Society’s International Registry, only 75% had evidence of hemophagocytosis at diagnosis. In some cases, repeated bone marrow analyses may be necessary to document hemophagocytosis. Neither the pathophysiologic basis for hemophagocytosis nor its precise mechanism is well understood. In HLH, hemophagocytosis is a reactive process that presumably results from the dysregulated immune response.
Related posts:
 The Neonatal Erythrocyte and Its Disorders
The Neonatal Erythrocyte and Its Disorders
 Diagnostic Approach to the Anemic Patient
Diagnostic Approach to the Anemic Patient
 Approach to the Child with a Suspected Bleeding Disorder
Approach to the Child with a Suspected Bleeding Disorder
 Disorders of the Red Cell Membrane
Disorders of the Red Cell Membrane
 Pediatric Myeloid Leukemia, Myelodysplasia, and Myeloproliferative Disease
Pediatric Myeloid Leukemia, Myelodysplasia, and Myeloproliferative Disease
 Nonrhabdomyosarcoma Soft Tissue Sarcomas and Other Soft Tissue Tumors
Nonrhabdomyosarcoma Soft Tissue Sarcomas and Other Soft Tissue Tumors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


