Chapter Outline
GENETIC PREDISPOSITION AND LIVER TUMORIGENESIS
Familial Adenomatous Polyposis
Overgrowth Syndromes and Hepatoblastoma
Genetic Syndromes and Hepatocellular Carcinoma
ENVIRONMENTAL EXPOSURES AND HEPATOBLASTOMA
VIRAL HEPATITIS AND LIVER TUMORS IN CHILDREN
PRESENTATION AND EVALUATION OF THE CHILD WITH A LIVER MASS
PATHOLOGY OF HEPATOBLASTOMA AND HEPATOCELLULAR CARCINOMA
ACQUIRED GENETIC CHANGES IN HEPATOBLASTOMA
TREATMENT: CHEMOTHERAPY IN HEPATOBLASTOMA
TREATMENT OF HEPATOCELLULAR CARCINOMA
SURGICAL ASPECTS OF LIVER TUMORS
LESS COMMON MALIGNANT TUMORS OF THE LIVER
Introduction and Epidemiology
Liver tumors in children account for approximately 1.1% of all malignancies in children younger than 20 years of age according to the National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) reports. The age-adjusted incidence rate of liver tumors is approximately 11 per million in infants younger than the age of 1 year and approximately 6 per million in children between 1 and 4 years of age. This translates to approximately 100 to 150 new cases of liver cancer annually in children in the United States.
Liver malignancies in children can be divided into three broad classifications: hepatoblastoma, which accounts for two thirds of malignant liver cancers and the predominant malignant tumor type in young children; hepatocellular carcinoma (HCC), the predominant tumor type in adolescents; and other rare hepatic malignancies of childhood. A spectrum of benign tumor histologies broadens the differential diagnosis of the infant or child with a liver mass.
In an early meta-analysis of eleven separate series totaling 1256 primary liver tumors in children reported by Weinberg and Finegold, 43% were hepatoblastoma, 23% HCC, 13% benign vascular tumors, 6% mesenchymal hamartomas, 6% sarcomas, 2% adenomas, 2% focal nodular hyperplasia, and 5% other tumors. Rare tumors of the liver also include rhabdoid tumors and hepatic germ cell tumors. An international working group recently provided guidelines regarding the histologic classification of pediatric liver tumors.
Most liver tumors in childhood are more common in boys than girls. The male-to-female ratio observed for hepatoblastoma in cooperative group trials has ranged from 1.5 to 1 to 2 to 1; a combined analysis of multiple studies suggests that the male-to-female ratio in hepatoblastoma is 1.65, consistent with SEER data.
Hepatoblastomas have a unique age distribution ( Fig. 58-1 ). Two peak ages occur, one at birth or within the first month of life and a second broad peak between 16 and 20 months of age. Ninety percent of hepatoblastomas occur in children younger than 4 years of age, and 95% of hepatoblastomas occur by age 5. Although rare, hepatoblastoma has been seen in adults, as described in multiple sporadic case reports. A systematic review of hepatoblastoma in adults suggests that these tumors are associated with a particularly poor prognosis, although the time frame over which these cases were gathered spanned multiple decades. In contrast hepatoblastomas diagnosed very early in life (i.e., in the neonatal period) are associated with a favorable outcome.
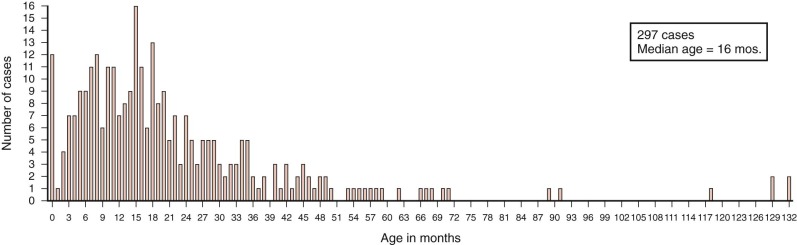
Hepatoblastoma in children older than 5 years of age is often more aggressive than the typical hepatoblastoma, has characteristics of HCC, and has been termed transitional liver cell tumor. More recently pathologists prefer to designate this poorly understood entity as hepatocellular tumor, not otherwise specified, pending a much-needed and more thorough understanding of the biology of these tumors with features of both hepatoblastoma and HCC. HCCs make up the bulk of liver tumors seen after age 10 years and throughout adolescence and adulthood. As with hepatoblastoma, HCC is more common in male subjects than female subjects. The fibrolamellar variant of HCC is a notable exception, with equal occurrence in male and female subjects.
The incidence of liver tumors has increased over recent decades, perhaps more than any other type of childhood tumor. During a single decade the incidence rates of hepatic tumors in infants younger than 1 year of age increased from 4 to 8 per million. The increase in rates was especially pronounced in female infants, in whom rates of hepatic cancer increased from 2 to 12 per million in a single decade. Ross and Gurney estimated that the average annual percentage change in incidence of hepatoblastoma between 1973 and 1992 was 5.2% for males and 8.2% for females. The rise in incidence of hepatoblastoma has been estimated to be 4% per year between 1992 and 2004 and continues to rise in recent years compared with even recent SEER data. The largest contributing factor to the increased incidence is thought to be the increased survival rates of premature infants who are at markedly increased risk of hepatoblastoma, as discussed in subsequent sections of this chapter.
Origins of Hepatoblastoma
Hepatoblastoma has long been thought to have prenatal origins, and increasing evidence points to prematurity, prenatal exposures, and overgrowth in early infancy as factors contributing to hepatoblastoma. It has long been presumed that embryonal tumors derive from primitive cells and that hepatoblastoma, as an embryonal tumor of the liver in which cells morphologically resemble cells in the developing embryonal and fetal liver, derives from a primitive hepatic cell. In rodents immature oval cells that proliferate after exposure to hepatocarcinogens express both biliary and hepatocytic markers. These cells are characterized by staining with OV-1 and OV-6 markers. Ruck and colleagues demonstrated the existence of a similar cell type in hepatoblastoma on the basis of marking with OV-1 and OV-6 in a small population of cells within the tumor. These OV-1 and OV-6 staining cells account for fewer than 1% of the cells within the tumor and provided evidence to support the existence of a population of stem cells in hepatoblastoma. Fiegel and coworkers analyzed hepatoblastomas for stem cell markers including CD34, Thy1, and c-kit and demonstrated the co-occurrence of these stem cell markers with hepatobiliary markers in ductal cells phenotypically resembling stem cells, which suggests that stemlike cells play a role in the histogenesis of hepatoblastoma. Other markers of hepatic progenitor cells including epithelial cell adhesion molecule (EpCAM), cytokeratin 19 (CK19), octamer-binding transcription factor 3/4 (Oct-3/4), and delta-like 1 homologue (DLK1) have been shown to be expressed in hepatoblastoma tumor cells. NOTCH2, which peaks in the second half of fetal development and then declines toward birth, is expressed in most hepatoblastomas. CITED1, expressed in embryonic mouse liver and a marker of hepatic progenitor cells, is shown to be present in hepatoblastoma, providing another similarity of hepatoblastoma to hepatic precursor stem cells.
Genetic Predisposition and Liver Tumorigenesis
Most cases of childhood liver malignancy are not associated with an apparent major genetic syndrome. However numerous syndromes have been observed in association with hepatoblastoma or HCC, as well as some benign hepatic tumors. A complete summary of genetic syndromes and liver tumors in children is provided in Table 58-1 . The major genetic syndromes associated with childhood liver tumors are discussed in the subsequent sections.
| Disease | Tumor Type | Gene |
|---|---|---|
| Familial adenomatous polyposis | Hepatoblastoma, adenoma, hepatocellular carcinoma, biliary adenoma | APC |
| Beckwith-Wiedemann syndrome | Hepatoblastoma, hemangioendothelioma | Multiple candidates |
| Simpson-Golabi-Behmel syndrome | Hepatoblastoma | GPC3 |
| Trisomy 18 | Hepatoblastoma | No single gene |
| Li-Fraumeni syndrome | Hepatoblastoma, undifferentiated sarcoma | TP53 |
| Glycogen storage disease types I-IV | Hepatocellular adenoma, carcinoma hepatoblastoma | Glucose-6-phosphatase |
| Hereditary tyrosinemia | Hepatocellular carcinoma | Fumarylaceto-acetate hydrolase |
| Alagille syndrome | Hepatocellular carcinoma | JAGGED-1, NOTCH2 |
| Other familial cholestatic syndromes | Hepatocellular carcinoma | FIC1, ABCB11 |
| Neurofibromatosis | Hepatocellular carcinoma, malignant schwannoma, angiosarcoma | NF-1 |
| Ataxia—telangiectasia | Hepatocellular carcinoma | ATM |
| Fanconi anemia | Hepatocellular carcinoma, fibrolamellar cancer, adenoma | FAA, FAC, others (20%) |
| Tuberous sclerosis | Angiomyolipoma | TSC1, TSC2 |
Familial Adenomatous Polyposis
Familial adenomatous polyposis (FAP) is caused by germline mutation of the APC gene, FAP is best characterized by the presence of colonic polyps, which without intervention are associated with the virtual certainty that adenocarcinoma of the colon will develop. Germline mutation of the APC gene is, however, also associated with a marked increased risk of hepatoblastoma. The risk of hepatoblastoma in children in FAP kindred is estimated to be 800-fold. The risk that a parent with an APC mutation will have a child with hepatoblastoma is 0.4%. Because FAP is an autosomal dominant disorder, genetic testing should demonstrate a predisposing mutation in 50% of offspring, such that the risk of hepatoblastoma in an infant who is a documented mutation carrier would be slightly less than 1%. Within a series of consecutive hepatoblastoma patients for whom family histories were obtained, approximately 8% of sequential cases of hepatoblastoma have a family history suggestive of FAP, and abnormalities of the APC gene were detectable in all cases.
Also reported in children from FAP kindreds, albeit less commonly, are other liver tumors, including HCCs. A benign precursor lesion, hepatocellular adenoma has been reported in a patient with a germline APC mutation and colonic polyps. Within resected liver tissue from children with germline APC mutations and hepatoblastoma, multiple hepatic nodules were consistent with precursor lesions with potential to progress to either hepatoblastoma or HCC. Presumably the timing of additional mutations or key developmental processes could determine the resulting histologic tumor type in the predisposed liver.
Significantly of nine reported cases of hepatoblastoma in siblings, at least four were in families with FAP; Of the remaining reported sibling pairs, one was affected with type 1a glycogen storage disease, and two sibling pairs had no apparent familial syndrome, and one recent report of a sibling pair with hepatoblastoma demonstrated an APC mutation in only one of the siblings. These reports suggest that whereas APC mutation accounts for a significant percentage of familial cases of hepatoblastoma, additional predisposition genetic changes may exist either as a distinct underlying cause of hepatoblastoma or as a modifying factor in infants with an existing APC mutation.
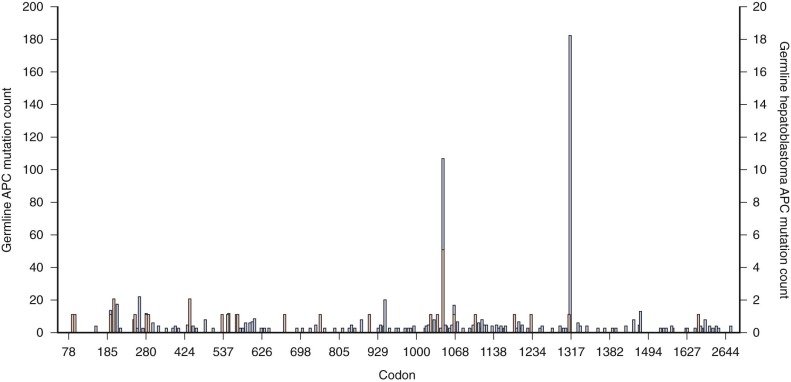
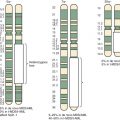
The percentage of sporadic hepatoblastoma patients (those without an obvious family history) who carry a germline mutation of the APC gene is not well defined. However it has been suggested both that children with hepatoblastoma be screened for germline APC mutations and that asymptomatic children in families with known polyposis be screened for hepatoblastoma. Within the APC gene genotype-phenotype correlations suggest regions of the gene that predict both the severity of polyposis as well as the presence of extracolonic manifestations. The literature suggests that germline mutations in FAP families with children who are affected by hepatoblastoma can occur throughout the gene ( Fig. 58-2 ) and can also include whole gene deletions or chromosome rearrangements such that the site or type of the individual APC mutation cannot be used to predict the occurrence of hepatoblastoma. This finding implies a risk to all APC mutation carriers.
Overgrowth Syndromes and Hepatoblastoma
Beckwith-Wiedemann syndrome (BWS) is an overgrowth disorder classically characterized by large birth weight, macroglossia, omphalocele, and visceromegaly. It is associated with a markedly increased risk of all embryonal tumors, including Wilms tumors, hepatoblastoma, neuroblastoma, and adrenal cortical adenoma. A registry of children with BWS followed at the National Cancer Institute determined that the relative risk of hepatoblastoma in children with BWS was 2280, higher than the relative risk of any other type of cancer in BWS. Unlike hepatoblastoma and FAP, hepatoblastoma has not been seen in siblings with BWS, despite the extraordinarily high relative individual risk in a patient with BWS. Presumably, this is because of the low incidence of familial occurrence of BWS.
BWS maps to chromosome 11p15, a region known to show loss of alleles in hepatoblastoma as well as parental-specific expression of genes (i.e., imprinting), including IGF2 and H19 . Molecular subtyping demonstrated that genetic alterations underlying BWS include mutations, epigenetic events involving imprinted genes, or uniparental disomy and that imprinting defects are of two types—those with imprinting defect at the imprinting control element 1 (IC1) at distal 11p15.5 and involving IGF2 and H19 hypermethylation and those with a defect at the imprinting control element (IC2), which is more proximal and involved imprinting changes of KvDMR1 and KCN11OT1. One series demonstrated that significantly more neoplasia is noted with either uniparental disomy or imprinting defects at IC1, although hepatoblastoma has been observed in BWS patients with mutations at the IC2 locus. Although differences may exist specific to the type of underlying genetic alteration, it is not possible at this time to tailor screening recommendations to genetic subtype. Present screening recommendations involve periodic ultrasonography of the abdomen and measurement of serum alpha-fetoprotein (AFP). In a series of tumors detected in children with BWS who underwent such surveillance, all tumors were small and resectable at diagnosis. Because hepatoblastoma has been noted to occur in the early neonatal period in BWS, some experts recommend screening at an early age. Even though AFP levels are normally very high in the neonatal period, screening with AFP should confirm a downward trend in the absence of tumor.
Simpson-Golabi-Behmel syndrome (SGBS) is an X-linked syndrome characterized by overgrowth with clinical features reminiscent of BWS, including an increased risk of embryonal tumors. Individual cases of SGBS with hepatoblastoma have been reported. The underlying gene for SGBS, GPC3, is highly expressed in hepatoblastoma compared with normal liver, which is consistent with a potential role in the tumorigenesis of hepatoblastoma. Although no longitudinal tumor surveillance studies have been carried out for SGBS, the overlap in tumor types observed suggests that surveillance similar to that used for BWS may be appropriate.
Sotos syndrome, an overgrowth syndrome that has clinical features overlapping with BWS and is associated with a predilection for tumors, has been reported in a child with hepatoblastoma. No large series have been reported, however, and the benefit of screening is unclear at this time.
Other Syndromes
Li-Fraumeni syndrome predisposes affected individuals to multiple tumor types in childhood and early adulthood. Although liver tumors are not typically included in Li-Fraumeni syndrome, hepatoblastoma has been reported in two Asian Li-Fraumeni kindreds. A kindred has also been reported in which a child developed undifferentiated (embryonal) sarcoma of the liver. Acquired mutations of the TP53 gene in hepatoblastoma tumor specimens, however, are rare or absent but have been reported in the undifferentiated embryonal sarcoma of the liver.
Both hepatoblastoma and HCC have occurred in children with primary biliary atresia. However it is unclear whether liver tumor development in children with biliary atresia is a complication resulting from liver disease or whether a common predisposition to both biliary atresia and tumor development exist. Significantly the same immature cells thought to share similarities to stem cells seen in hepatoblastoma are also seen in biliary atresia. Multiple cases of hepatoblastoma and trisomy 18 have been reported. This is somewhat notable in that trisomy of chromosome 18 is not seen as an acquired event despite the observation of multiple other recurring trisomies.
Hepatoblastoma has been reported in a female subject with monosomy 7 myelodysplasia, a predisposition syndrome not previously seen with hepatoblastoma. Hepatoblastoma has also been reported in neurofibromatosis type 1, although it is not characteristic of this disorder.
A recent report has noted a possible association of hepatoblastoma with prune-belly syndrome, a congenital disorder of lax abdominal musculature and urogenital abnormalities. A review of data from the Children’s Oncology Group (COG) demonstrated a significant association with kidney and bladder congenital abnormalities.
Genetic Syndromes and Hepatocellular Carcinoma
HCC, in contrast to hepatoblastoma, is seen in genetic syndromes that are characterized by hepatic cirrhosis. Progressive familial intrahepatic cholestasis, a condition marked by neonatal hepatic cirrhosis, is associated with liver tumor development in children. Both HCC and cholangiocarcinoma are known to occur in this disorder. The disorder, also known as Byler disease, is a heterogeneous syndrome. The underlying genetic etiology is a germline mutation of the ABCB11 gene, which encodes a bile salt export pump. In a series of 11 cases of HCC in children younger than 5 years with neonatal hepatitis with clinical characteristics of familial intrahepatic cholestasis, 10 had immunohistochemical evidence of a deficiency in bile acid transporter bile salt export pump. In all patients in whom genotyping was possible, a germline mutation of the ABCB11 gene encoding bile salt export pump was detected.
Alagille syndrome is an autosomal-dominant disorder characterized by a characteristic facies, congenital heart disease, intrahepatic cholestasis with neonatal jaundice, and a paucity of hepatic bile ducts resulting in neonatal jaundice. The liver pathology may progress to frank cirrhosis. HCC has been reported in children with Alagille syndrome. The vast majority of these Alagille syndrome–associated HCCs occur in boys. In one family three children were reported with HCC in the absence of any other type of liver disease other than the Alagille syndrome. Alagille syndrome is associated with germline mutations in the JAGGED1 gene, the endogenous ligand for NOTCH2, a developmental gene involved in controlling maturation of the heart and liver. In a minority of cases of Alagille syndrome, in which the JAGGED1 gene is not found to be mutated, the NOTCH2 gene itself is mutated. It has been shown that the mechanism of liver disease in Alagille syndrome may relate to an increased expression in hepatocyte growth factor in cells that carry JAGGED1 mutations.
The family of glycogen storage disorders has been associated with liver tumorigenesis. Glycogen storage disease type I has been reported in association with benign adenomas, HCC, and hepatoblastoma, although the latter has been reported only in adolescents. Similarly glycogen storage disorder type III has been associated with HCC, and type IV has been associated with hepatocellular adenoma. Because HCC is known to occur in diseased liver, with long-term follow-up HCC may be considered a complication of increased survival of patients with glycogen storage disease.
Hereditary tyrosinemia type 1, characterized by the deficiency of fumarylacetoacetate hydrolase is likewise associated with HCC in the setting of severe liver disease.
Fanconi anemia is associated with liver tumors, although the association has been almost always reported in association with the use of androgenic steroid treatment. In a review of 1301 reported cases of Fanconi anemia, 2.8% of subjects were reported to have a liver tumor. The most common type of liver tumor reported in Fanconi anemia is HCC, but adenomas are also seen in these patients. The incidence appears to increase with age and does not appear to plateau. In multiple instances liver malignancy was seen in patients with Fanconi anemia who also had a diagnosis of leukemia. The extent of the associated androgen treatment with development of hepatic malignancy versus the effect of the underlying genetic defects in DNA repair as manifested by chromosome breakage in Fanconi anemia is unclear, but a review of the association of androgens and liver tumors suggests that patients with Fanconi anemia develop liver tumors after smaller and briefer androgen exposure than do individuals without Fanconi anemia. It is also unclear whether there is a difference among the genetic subtypes of Fanconi anemia with regard to the predisposition to hepatic and other malignancy.
Genetic Syndromes and Other Benign Liver Tumors
In addition to syndromes predisposing to malignant liver tumors, several syndromes are associated with benign liver tumors. Tuberous sclerosis has been linked to development of benign hepatic angiomyolipomas. Although these hepatic growths are not as common as renal angiomyolipomas, which may develop in as many as 80% of patients with tuberous sclerosis, the hepatic lesions are evident in imaging studies in approximately 13% of patients with tuberous sclerosis, according to one study.
Prematurity as a Risk Factor
One of the most intriguing aspects of hepatoblastoma is its association with premature or low-birth-weight infants. This association was initially reported by Japanese investigators who analyzed patient diagnoses and birth weights in the Japan Children’s Cancer Registry over the 9-year period from 1985 to 1993. It was found that 3.9% of patients with hepatoblastoma were of very low birth weight. Moreover it was observed that when data were subdivided over 4-year periods, the percentage of hepatoblastoma cases of low birth weight rose from 0.7% in 1985 to 1989 to 8.6% in 1990 to 1993 and demonstrated a linear increasing trend. The risk of hepatoblastoma has also been shown to be inversely proportional to birth weight. The relative risk for an infant between 2000 and 2999 grams is 1.21, whereas the relative risk for an infant of less than 1000 grams is 15.64.
Data from the Children’s Cancer Group survey confirmed the preponderance of former premature infants in a series of 76 patients with hepatoblastoma. A second confirmation in the United States comes from the California population-based cancer registry, which also reported an elevated risk of hepatoblastoma in children of very low birth weight. This study also pointed out that the age of diagnosis of hepatoblastoma in children of low birth weight was actually older than that of children of normal birth weight, which could partly be explained by gestational age.
It has been speculated that the increase in the percentage of low-birth-weight infants among children with hepatoblastoma may be partially caused by the increased survival rates of low-birth-weight infants. It is also believed that the increase in survival rates of premature infants at risk for hepatoblastoma may contribute to the overall increase in childhood liver tumors.
Indeed the rise in the rate of hepatoblastoma has been found to be consistent with the risk in births of low-birth-weight infants. However whether the pathways by which low-birth-weight infants develop hepatoblastoma involve key exposures in the newborn period, increased sensitivity of the liver in premature infants to potential carcinogens, disruption of the normal process of liver development by premature birth, or a combination of these factors has yet to be defined. One small single-institution study, in which treatment records of five infants who developed hepatoblastoma were compared with those of infants of similar birth weights who did not develop hepatoblastoma, suggests that perinatal intensive long-term treatments contribute to the development of hepatoblastoma. A somewhat larger study comparing 12 hepatoblastoma cases and 75 birth weight–matched controls suggests that the duration of oxygen therapy, use of furosemide, and length of time taken to regain body weight at birth were factors in predicting the likelihood that hepatoblastoma would develop in premature infants. A study from the United Kingdom reported that polyhydramnios and either eclampsia or preeclampsia are more common in children with hepatoblastoma compared with those without hepatoblastoma, and that the eclampsia or preeclampsia observed was also associated with low birth weight.
A recent case-control study of 600 COG patients with hepatoblastoma (AEPI04C1) has examined exposures in low-birth-weight and other infants with hepatoblastoma and has not found an association with maternal illness or medication use during pregnancy or infertility treatment and hepatoblastoma. Previously use of assisted reproductive technology was postulated to be a contributing factor, although it is thought that it may contribute to hepatoblastoma development only through the associated risk of BWS and assisted reproductive technology (ART), but not as an independent risk factor.
Environmental Exposures and Hepatoblastoma
An early epidemiologic case-control study of risk factors for hepatoblastoma by Buckley and colleagues, based on parental interviews, revealed an association of hepatoblastoma with maternal occupational exposure to metals, such as those used in welding or soldering (odds ratio [OR] = 8); petroleum products such as lubricating oils or greases (OR = 3.7); and paints or pigments (OR = 3.7). There was also a significant association with paternal occupational exposure to metals (OR = 3, P = .01) and a marginally significant association with paternal occupational exposure to petroleum products (OR = 1.9). Although the aforementioned study did not find an association of parental smoking with hepatoblastoma, this has been reported in several subsequent studies. The risk of hepatoblastoma in children of parents who smoked was approximately double in all three studies. Indeed in 2009 the International Agency of Research on Cancer declared that parental smoking is a carcinogen in the developing liver. Subsequent findings have been variable, however. A recent report from COG, the largest etiologic study to date, reported no association between smoking and hepatoblastoma. Variability among studies, however, could be based on small numbers and differences in reporting and overall smoking prevalence in the different populations. Although alcohol clearly plays a role in cirrhosis of the liver and HCC in adults, the COG study reported no association with parental alcohol use and hepatoblastoma in offspring.
Viral Hepatitis and Liver Tumors in Children
Viral hepatitis infection has historically accounted for a large percentage of HCC cases. Before the introduction of the hepatitis B vaccine in Taiwan, HCC accounted for 80% of cases of liver tumors in children. With the introduction of the hepatitis B vaccination in 1984, rates of liver tumors in Taiwanese children older than 6 years dropped significantly, from 0.70 per 100,000 from 1981 to 1986 to 0.36 per 100,000 between 1990 and 1994. The decrease in rates of HCC after introduction of the vaccine was more pronounced in boys than in girls. In fact the incidence of HCC decreased in boys but not in girls. The male-to-female incidence of HCC in Taiwan steadily decreased from 4.5 per 100,000 in 1981 to 1986 to 1.9 per 100,000 in 1990 to 1996, 6 to 12 years after the introduction of the vaccine. The reasons for these sex-related differences in responses to the vaccine are not known. It has been demonstrated that among children with hepatitis B the presence of a deletion of the pre-S region of the hepatitis B virus was an additional risk factor for the development of HCC (OR = 36.69).
Presentation and Evaluation of the Child with a Liver Mass
Aside from patients with known genetic syndromes (discussed in the preceding sections), most cases of hepatoblastoma occur in otherwise well children who present with an abdominal mass, often with some degree of abdominal discomfort.
Isosexual precocious puberty has been observed in some boys as a presenting finding in hepatoblastoma and occurs secondary to tumor production of chorionic gonadotropin. In one study of 48 children, the incidence of precocious puberty in children with liver tumors was 6%. To the authors’ knowledge, there are no known cases of precocious puberty associated with hepatoblastoma in girls.
Laboratory evaluation of the child with a liver mass should include a complete blood count and liver function tests, as well as AFP and β-human chorionic gonadotrophin tumor marker levels. The platelet count is often high in hepatoblastoma but is not diagnostic of hepatoblastoma among children with liver tumors. This thrombophilia is associated with high serum levels of thrombopoietin (TPO), also known as c-Mpl ligand. Normally synthesized in the liver, TPO has also been shown to be expressed in hepatoblastoma tumor tissues and is present at higher than normal levels in the serum of patients with hepatoblastoma. In a large series of patients of various ages (13 to 84 years) who had hepatic tumors, thrombocytosis was noted in 2.7%. The high platelet count was correlated with higher serum TPO levels. Other components of the complete blood count are usually normal insofar as bone marrow involvement with liver tumors has not been reported.
AFP is the primary serum marker that is used diagnostically and prognostically, as well as in surveillance. A sample should be obtained in the initial evaluation of any child with a liver mass. AFP levels are markedly elevated in more than 90% of hepatoblastomas and more than 50% of HCCs. Care should be taken in interpreting AFP levels in the young infant because levels are normally very high at birth and decline to less than 10 ng/dL over the first year of life. Of note is that some liver malignancies—notably the small cell undifferentiated subtype of hepatoblastoma as well as the fibrolamellar variant of HCC—are not associated with elevation of the AFP. Likewise, sarcomas and rhabdoid tumors are not associated with elevated AFP. The International Society of Oncology Epithelial Liver Group (SIOPEL) has demonstrated that hepatoblastomas with low AFP levels at diagnosis (less than 100 ng/mL) tend to present at a higher stage and are associated with a poor outcome. A low AFP level has also been noted to be a marker of poor outcome across multiple studies compiled and analyzed by the Children’s Hepatic Tumor International Consortium (CHIC) group. The β-human chorionic gonadotrophin level is only occasionally elevated in hepatoblastoma, usually in cases that present with precocious puberty. The β-human chorionic gonadotrophin level is also elevated in the rare choriocarcinoma of the liver, as well as teratomas.
Evaluation of a child with a liver tumor should include a thorough family history to document specific cancers in relatives, with notation made of any early-onset colon cancers or colectomies, in addition to the presence of thyroid cancers or medulloblastoma, which can be part of FAP syndrome. Testing for germline mutation of the APC gene may be indicated; if performed, it should include appropriate genetic counseling. These patients with hepatoblastoma and germline APC mutations will require long-term surveillance for colonic polyps and tumor development.
In addition the medical history should note the birth weight, history of prematurity, and presence or absence of stigmata of neonatal overgrowth, including omphalocele and hemihypertrophy, which would suggest BWS and a risk for other embryonal tumors.
Staging
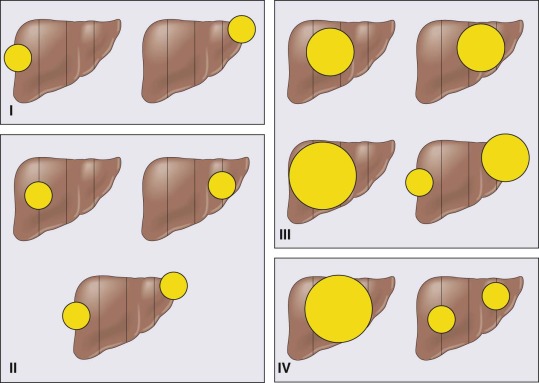
Although North American trials have traditionally used a postsurgical diagnostic staging group, applied after the initial surgical procedure (biopsy or resection), the European system, based on the appearance by imaging before chemotherapy and surgical resection (PRETEXT), is being adopted worldwide. PRETEXT stages I through IV reflect the number of sectors that are involved with tumor. Other components of the staging system reflect extension into the vena cava , extension into the portal vein, extrahepatic abdominal disease, or distant metastases. The SIOPEL staging is shown schematically in Figure 58-3 . A comparison of the prior North American staging and the current SIOPEL staging is shown in Table 58-2 .
| North American Staging | European Staging SIOPEL/PRETEXT | |
|---|---|---|
| Postsurgical Staging | Presurgical Staging | |
| Stage 1 | No metastases; tumor completely resected | Tumor involves only one quadrant; three adjoining liver quadrants free of tumor |
| Stage 2 | No metastases, tumor grossly resected with microscopic residual disease (i.e., positive margins, tumor rupture, or tumor spill at the time of surgery) | Tumor involves two adjoining quadrants; two adjoining quadrants are free of tumor |
| Stage 3 | No distant metastases, tumor unresectable or resected with gross residual tumor, or positive nodes. | Tumor involves three adjoining quadrants or two nonadjoining quadrants; one quadrant or two nonadjoining quadrants are free of tumor |
| Stage 4 | Distant metastases regardless of the extent of liver involvement. | Tumor involves all four quadrants; there is no quadrant free of tumor |
Figure 58-4 demonstrates a small hepatoblastoma tumor image that was initially diagnosed by surveillance ultrasound in a patient with BWS. This tumor involved two sectors and as such is classified as PRETEXT II, but because it was completely surgically excised after diagnosis, it would be stage 1 using the North American staging system. Figure 58-5 demonstrates a multifocal hepatoblastoma that involves all four sectors and had metastatic lesions in the lung; therefore it is PRETEXT IV by the North American staging system. If the patient had not had metastatic lesions, the tumor would be stage 3 according to the North American system but still PRETEXT IV.
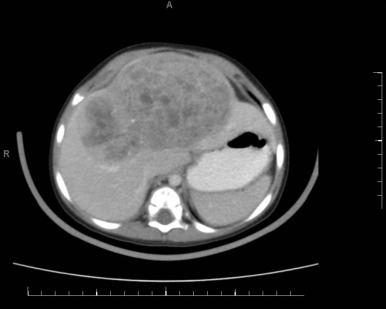
Both staging systems have been shown to have prognostic significance, but it is difficult to compare studies using different staging systems. In the future the PRETEXT system will be used by most groups internationally.
Pathology of Hepatoblastoma and Hepatocellular Carcinoma
Grossly hepatoblastomas occur as single expansile masses, usually in the right lobe of the liver. A series of international discussions originating at the Los Angeles COG International Pathology Liver Tumors Symposium in 2011 resulted in a consensus classification of liver tumors in children, summarized in Table 58-3 . A description of immunohistochemical properties currently recommended to aid in the classification of pediatric liver tumors is provided in Table 58-4 .
| Epithelial Tumors | Mesenchymal tumors |
|---|---|
|
|
|
|
| Fetal WDF | Fetal with Mitoses | Pleomorphic | Embryonal | Small Cell Undifferentiated | Mesenchymal | Cholangioblastic | |
|---|---|---|---|---|---|---|---|
| GPC3 | Finely granular | +++ Coarse | ++ Coarse | +++ Coarse/rare − | −, Rare+cell | − | − |
| β-cat | Variably +/+++ nuclear or normal | +/+++ Nuclear | +/+++ Nuclear | +/+++ Nuclear, can be− | +++ Nuclear | +++ Nuclear on osteoid/blastema/neg in teratoid elements | Variable/positive nuclei |
| GS | +++ | +++ | Variable | Variable, can be neg | − | − | − |
| Hep Par | +++ | +++ | Variable | Usually − | − | − | − |
| Cyclin D1 | − | +/++ | +/+++ | +/+++ | +/++ | Variable/neg in teratoid | |
| CK7 | − | − | − | −/+ | Variable/weak in blastema | +++ | |
| CK19 | − | − | − | − | +/++ Variable | Neg/weak in blastema | +++ |
| CD34 | + Endo | + Endo | |||||
| Vimentin | − | − | − | − | +/++ | +++ | Usually − |
| INI1 | +++ | +++ | +++ | +++ | Negative in pure SCU; variable when mixed | +++ | +++ |
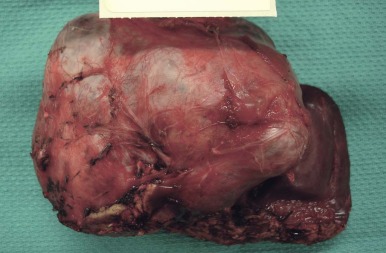
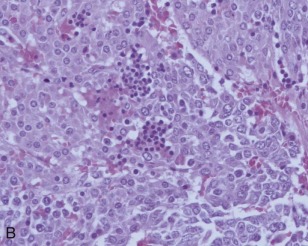
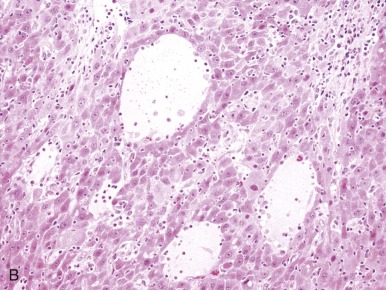
A representative resected hepatoblastoma tumor is shown in Figure 58-6 . Hepatoblastomas are composed primarily of epithelial cells that resemble different stages of the developing liver. Most hepatoblastomas are a mixture of cells resembling fetal cells and cells resembling embryonal cells. A typical hepatoblastoma of fetal and embryonal histology is shown in Figure 58-7, A . Because hepatoblastomas derive from immature cells that may retain the potential to differentiate, tumors often have foci of other types of more differentiated cells, including hematopoietic cells, as demonstrated in Figure 58-7, B . A subset of hepatoblastomas are characterized by cholangiocyte-like cells, which suggest the differentiation of the neoplastic cells along the cholangiocyte lineage as opposed to the hepatocyte lineage. When the epithelial cells are aligned in thick cordlike structures, the tumor is described as having macrotrabecular features. In approximately 5% of hepatoblastomas, only fetal-type cells are seen histologically, as shown in Figure 58-7, C . These fetal-only tumors are now further classified according to the degree of mitoses.
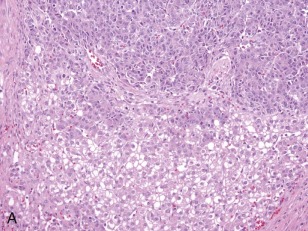
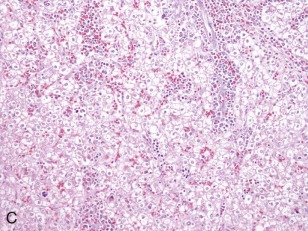
Two histologic subtypes of hepatoblastoma have significant implications for predicting clinical outcome. Approximately 7% of hepatoblastomas are composed of pure, well-differentiated fetal cells. The well-differentiated histology, previously referred to as pure fetal histology, has been associated with an exceptionally favorable outcome. A recent COG study by Malogolowkin and coworkers demonstrated that if the hepatoblastoma is completely resected surgically at the time of diagnosis, further therapy may be unnecessary. The presence of this subtype and the desire to avoid the toxicity of chemotherapy remain compelling reasons to pursue upfront resection whenever possible.
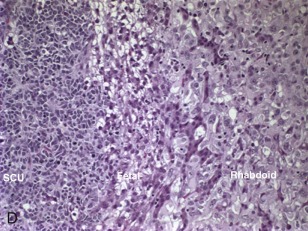
The small cell undifferentiated variant accounts for approximately 5% of hepatoblastoma and also carries clinical significance. Histologically the cells are small and pleomorphic and show high mitotic rates, as shown in Figure 58-7, D . There are several reports in small cell undifferentiated hepatoblastoma of unique chromosome translocations involving chromosome 22q12, the region of SMARCB/INI1, the gene most often associated with rhabdoid tumors and atypical teratoid tumors. Emerging evidence suggests that from histologic, cytogenetic, molecular genetic, and clinical perspectives these tumors resemble rhabdoid tumors to varying degrees. Immunohistochemistry demonstrated loss of the INI1 protein in the small cell undifferentiated variant, and it is now recommended that INI1 staining be performed as part of the histologic characterization (see Table 58-4 ). Mounting evidence suggests that the small cell variant is associated with a poor outcome.
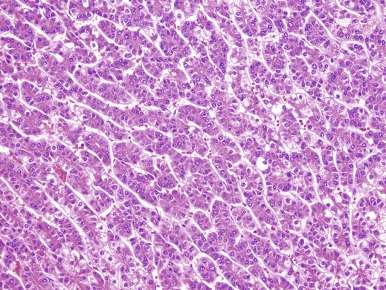
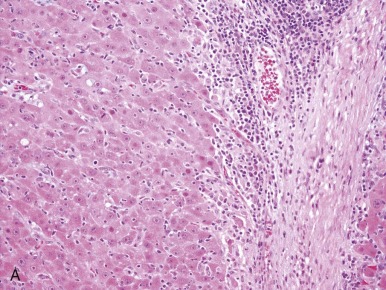
HCCs consist of cells that are well-differentiated and resemble normal hepatocytes. A typical HCC is shown in Figure 58-8 . The fibrolamellar variant of HCC is characterized by larger, polygonal cells mixed with fibrous stroma and often eosinophilic cytoplasm, occasionally with cystic degeneration ( Fig. 58-9 ). In contrast to hepatoblastoma, the adjacent liver tissue in patients with HCC is often marked by cirrhosis. The fibrolamellar variant is an exception in which surrounding liver tissue is usually noncirrhotic.
Acquired Genetic Changes in Hepatoblastoma
Karyotypic changes in hepatoblastomas can be classified into two broad categories: numeric changes and structural changes of individual chromosomes, the most common of which involve translocations of chromosome 1. The initial recurring translocation described was the translocation involving chromosomes 1 and 4, t(1;4)(q12;q34). It has since been reported in a large series that hepatoblastoma is characterized by a family of chromosome translocations with similar breakpoints on either chromosome 1q12 or 1q21. Each such translocation observed is unbalanced, resulting in a gain of the long arm of chromosome 1, and is also frequently associated with numerous whole chromosomal gains.
Most of the numeric aberrations result in addition of whole chromosomes, but occasionally they result in loss of chromosomes. The specific numeric changes are nonrandom. Trisomies of chromosome 2, 8, and 20 are the most common recurring numeric aberrations. The most commonly observed chromosomal losses are chromosomes 4 and 18. Although these whole chromosome changes have been described previously by classical karyotype analysis, they can perhaps be better visualized by whole genome comparative genomic hybridization, as shown in Figure 58-10 . The clinical significance of these chromosomal abnormalities is not yet known..
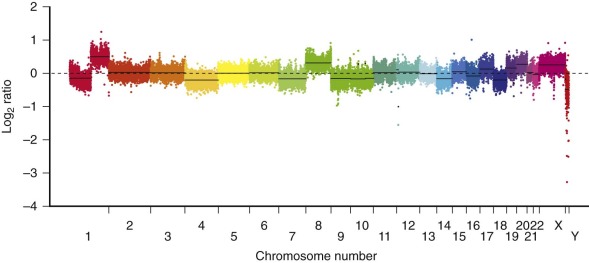
HCC is also characterized also by chromosomal gains and losses, with loss of the Y chromosome a notable finding. Duplication of chromosome 1q is also seen in HCC with increased expression of numerous chromosome 1 genes. Cytogenetic data on the fibrolamellar variant of HCC is sparse, but one childhood fibrolamellar carcinoma has been characterized by a hypertriploid karyotype with clonal evolution and multiple additional chromosomal gains as well as loss of the Y chromosome.
Related posts:
 The Neonatal Erythrocyte and Its Disorders
The Neonatal Erythrocyte and Its Disorders
 Diagnostic Approach to the Anemic Patient
Diagnostic Approach to the Anemic Patient
 Approach to the Child with a Suspected Bleeding Disorder
Approach to the Child with a Suspected Bleeding Disorder
 Disorders of the Red Cell Membrane
Disorders of the Red Cell Membrane
 Pediatric Myeloid Leukemia, Myelodysplasia, and Myeloproliferative Disease
Pediatric Myeloid Leukemia, Myelodysplasia, and Myeloproliferative Disease
 Nonrhabdomyosarcoma Soft Tissue Sarcomas and Other Soft Tissue Tumors
Nonrhabdomyosarcoma Soft Tissue Sarcomas and Other Soft Tissue Tumors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


