Hemorrhagic Disorders: Introduction
Although hemorrhage caused by intracranial, aneurysmal, and gastrointestinal pathologies account for a significant proportion of all deaths in the older-than-60-year age group, fatal bleeding caused by primary defects in coagulation and platelets are less common. Nevertheless, the development of a bleeding diathesis in an elderly patient presents clinical challenges that form the focus of this chapter.
Hemostasis is a highly regulated process involving a dynamic balance between coagulation and fibrinolytic proteins and their inhibitors, platelets, and the vascular endothelium. The overall aim of the coagulation system is to rapidly generate a hemostatic plug at sites of vascular injury to prevent catastrophic blood loss from severed vessels. Congenital or acquired disturbances of the delicate balance between coagulation and fibrinolysis may result in thrombotic or hemorrhagic clinical manifestations.
Elderly patients more commonly present with acquired bleeding disorders, but occasionally a mild congenital bleeding diathesis may remain hidden until later years when surgical procedures become common, when comorbid disease develops (such as renal or hepatic dysfunction), or when an additional acquired condition creates an additive challenge to the hemostatic system, such as the introduction of antiplatelet agents or anticoagulant therapy. Focusing on bleeding in the elderly, this chapter (a) provides a brief overview of hemostatic mechanisms, (b) discusses the clinical assessment of an elderly patient with a history of bleeding, (c) details the main inherited and acquired hemorrhagic conditions and their presentation in an elderly population, and (d) outlines the treatment of elderly patients with hemorrhagic states.
Overview of Hemostasis
Following damage to the endothelium, blood is exposed to subendothelial components, such as von Willebrand factor (vWF) and collagen. Platelets adhere to the subendothelium and are activated, releasing substances that further recruit and activate platelets inducing platelet aggregation. A platelet plug is formed at the site of injury providing initial arrest of bleeding, in a process known as “primary hemostasis”. A process referred to as “secondary hemostasis” follows to ensure stabilization of the platelet plug.
Vascular injury also exposes tissue factor (TF), a membrane-bound procoagulant factor that triggers the coagulation cascade. The coagulation cascade is a highly regulated series of enzymatic reactions involving the sequential conversion of proenzymes into their active forms. Activated platelets provide a phospholipid surface membrane on which coagulation enzymes localize and assemble into complexes that accelerate the serial enzymatic reactions. Such amplification processes translate a small initiating signal into an explosion of thrombin generation, thereby resulting in rapid fibrin formation. The polymerization of fibrin stabilizes the platelet plug and results in secondary hemostasis.
Removal of the fibrin clot takes place by activation of the fibrinolytic system. Plasminogen, a zymogen, is converted into plasmin, an enzyme that degrades the fibrin matrix of the clot into soluble fragments. Hemostatic mechanisms rely on dynamic interactions between platelets, the coagulation cascade, the fibrinolytic system, and the endothelium (see Figure 106-1). Disturbances of the equilibrium, caused by acquired or congenital factors, may result in unexpected or excessive bleeding or thrombosis.
Figure 106-1.
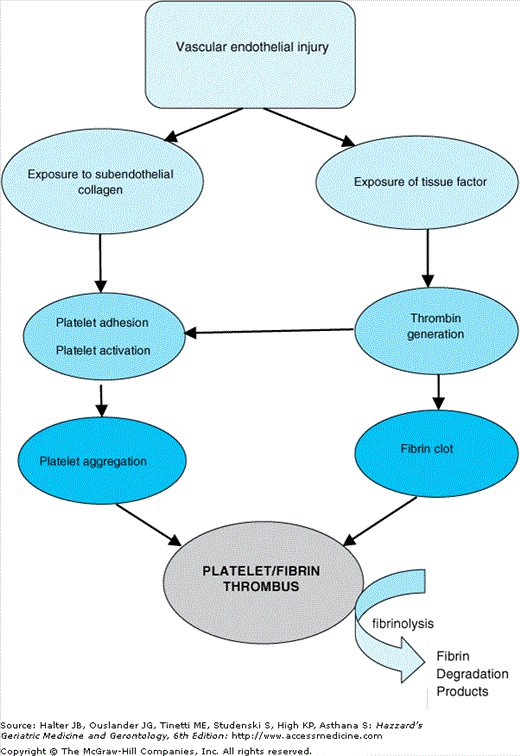
The hemostatic mechanism involves a dynamic relationship between the vascular endothelium, platelets, coagulation cascade, and fibrinolysis. Abnormalities of any parts of the system can cause a bleeding diathesis. (Modified with permission from Liaw PC, Weitz JI. Coagulation overview. Section 8. Hematologic problems. In Albert RK, Slutsky A, Ranieri M, Takala J, Torres A, eds. Clinical Critical Care Medicine. Philadelphia: Elsevier; 2006:543–553).
Two large epidemiological studies of men and women aged 25 to 74 years (the MONICA Project), and 60 to 79 years, demonstrate a significant effect of the ageing process on coagulation factors, inhibitors, and activation markers. However, these age-related changes do not cause an increased tendency to hemorrhage; instead, increases in fibrinogen and coagulation factors VII (fVII), VIII (fVIII), IX (fIX), and vWF are greater than increases in the coagulation inhibitors, antithrombin (AT), protein C, and protein S, resulting in a tendency toward thrombosis. This net procoagulant effect appears to be more pronounced in men than women, and is reflected by an increase in thrombin–AT complexes, high plasma levels of activation peptides cleaved from prothrombin, fIX, and factor X (fX), and minor changes in thromboelastograph variables. Levels of plasminogen activator inhibitor-1 (PAI-1), an inhibitor of the tissue plasminogen activator (tPA), also increase in an age-dependent manner, adding to the relative prothrombotic state with ageing.
Megakaryopoiesis does not appear to alter with the aging process. Platelets are present in similar numbers with only a normal or moderate reduction in life span compared with platelets in younger individuals.
Clinical Approach to Bleeding in the Elderly Patient
A patient may present for evaluation of new symptoms of bleeding or bruising, or may be referred postoperatively or postprocedurally following difficulty securing hemostasis. The immediate assessment of a patient who has been bleeding intraoperatively can be particularly challenging from a laboratory viewpoint, and reassessment may be necessary several weeks later once the hemostatic system has had time to rebalance. Accurate historical details are essential in the overall evaluation process (see Table 106-1). Accompanying caregivers are often helpful in providing extra details about patients who cannot provide a reliable history.
HISTORICAL DETAILS | POINTS TO ELICIT |
|---|---|
When did the bleeding start? | Duration of symptoms |
Lifelong or recent onset? | |
Nature of the bleeding? | Spontaneous or post trauma/procedure? |
Epistaxis, gingival bleeding | |
Previous history of menorrhagia | |
Joint bleeds, muscle bleeds | |
How long did the bleeding persist? | |
Was the bleeding immediate or delayed? | |
Any other associated forms of bleeding? | Gastrointestinal, hematuria |
Response to past operative or dental challenges | Dental extraction is a hemostatic challenge |
Medications | Include all recent medications |
Include herbal remedies | |
Diet | Any history of malabsorption? |
Recent administration of antibiotics? | |
Balanced diet? | |
Family history | Are male and female members of the family equally affected? |
Other questions | Any comorbidity to exacerbate a bleeding tendency? |
Does the bleeding require urgent attention or is this an evaluation to assess the potential to bleed during future operations? |
Bleeding issues that are lifelong are likely to hold a congenital basis, rather than a recently acquired etiology. An assessment of previous hemostatic challenges should be made, such as dental surgery, trauma, and operations, including operations commonly performed in childhood such as tonsillectomy and adenoidectomy. Bleeding around the time of childbirth may be more difficult to interpret, but the need for blood transfusion is valuable information that may point to the severity of hemorrhage. Congenital causes may manifest late in life if they are mild and the patient has not experienced any previous hemostatic challenges.
The nature of bleeding symptoms may determine the etiology of the hemostatic defect: mucosal bleeding, epistaxis, and menorrhagia highlight defects in platelets or their interaction with the vascular wall; delayed bleeding, spontaneous hemarthroses, and muscle bleeds are consistent with coagulation factor deficiencies such as hemophilia. The significance of symptoms such as bruising, epistaxis, and menorrhagia may be challenging to define as there is a strong subjective component to each, and they are common symptoms. Sometimes it is useful to assess for any temporal changes in severity of these events. The clinician should also determine whether bleeding occurs spontaneously, or secondary to trauma or surgery.
The term “purpura” refers to a spontaneous extravasation of blood from the capillaries into the skin. “Petechiae” are purpuric lesions of pin-head size and ecchymoses are larger lesions.
The location and extent of bruising may be indicative of an underlying bleeding disorder. For example, large ecchymoses, presenting on the trunk without known trauma, may suggest an underlying bleeding disorder, whereas minor bruising on extremities are more likely benign. In an older person with no prior history of bleeding, extensive or truncal ecchymoses may suggest the development of an acquired coagulation disorder such as liver disease, disseminated intravascular coagulation (DIC), or acquired hemophilia.
Tonsillectomy, adenoidectomy, and dental extraction of the molar teeth create a significant hemostatic challenge, and the patient’s response to the challenge is of historical importance; bleeding that required resuturing or excessive packing, or that continued for several hours to days is suggestive of an underlying hemostatic disorder.
This complaint is highly subjective and poorly correlates to actual blood loss; description of the passage of clots, a history of past iron-deficiency anemia, the requirement for blood transfusion or for hysterectomy at a young age may all highlight an underlying bleeding diathesis.
These symptoms are usually caused by localized blood loss from underlying pathology; an acquired or congenital bleeding disorder may unmask the symptoms and make them more florid. In an older person, such bleeding should prompt urgent investigations for the underlying cause as bleeding from such sites could be potentially life-threatening and may be a harbinger of malignancy or other serious conditions.
A patient’s first hemostatic challenge may occur in older age, leading to the presentation of a hidden mild congenital disorder. A careful family history should record any hemorrhagic events affecting family members, including immediate and more distant relatives. However, one-third of cases of hemophilia A and B arise as a result of de novo mutations in whom there may be no family history. Female carriers of factor VIII (fVIII) or factor IX (fIX) deficiency, or male patients with mild hemophilia may have no bleeding history if coagulation factor levels are greater than 30% to 50%; lower baseline factor levels may cause bleeding problems after surgical or dental challenge.
A careful history should be taken of all medications and herbal remedies (see Table 106-2) being administered. Aspirin, a cyclo-oxygenase-1 inhibitor, and ADP-receptor antagonists such as clopidogrel and ticlopidine cause platelet dysfunction; quinine, carbimazole, and penicillins may cause drug-induced immune thrombocytopenia (d-ITP) and anticoagulants including heparin, low-molecular-weight heparin, and warfarin may all cause hemorrhagic complications in elderly patients. The chronic administration of steroids may thin the skin increasing the likelihood of purpura (steroid-induced purpura). It may be difficult to solicit an accurate history of all medications used, so patients should be encouraged to bring all medications and dietary supplements with them to their consultation. It is also important to review the doses and frequency of administration.
MEDICINE | ACTION | COMMENTS |
|---|---|---|
Garlic | Inhibition of platelet aggregation; increased fibrinolysis | May increase bleeding when combined with other antiplatelet drugs |
Gingko | Inhibition of platelet activation | May increase bleeding when combined with other antiplatelet drugs |
Ginseng | Increases PT and aPTT | May increase bleeding risk; also reduces anticoagulant effect of warfarin |
St John’s Wort | Induction of cyt P450 enzymes | Interacts with warfarin |
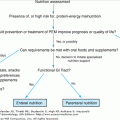
In an elderly patient presenting with new onset bleeding, an assessment of nutritional status is important. The most common dietary deficiencies that contribute to bleeding or a coagulopathy are those of vitamin B-12, folate, vitamin K, and vitamin C.
Vitamin B-12 and folate are essential for normal hematopoiesis because of their roles in DNA synthesis. Although vitamin B-12 or folate deficiency may cause megaloblastosis and pancytopenia, including thrombocytopenia, these conditions alone are unlikely to present with a bleeding diathesis unless the patient is taking concurrent anticoagulants or antiplatelet medications, or other comorbidity is present. Nevertheless, it is important to exclude vitamin B-12 and folate deficiency in patients presenting with thrombocytopenia or other cytopenias as it is easily correctable.
Acute folate deficiency can develop quite rapidly, within a 3-week period; this may be found in up to one-third of hospitalized patients. Risk factors include anorexia, acute alcohol ingestion, and in patients receiving anticonvulsants. Rich sources of dietary folate include green leafy vegetables, fresh and minimally cooked, as folate is thermolabile and is destroyed following prolonged cooking.
Vitamin B-12, a cobalamin, is synthesized by microorganisms, and must be solely obtained from the diet; foods rich in vitamin B-12 include meat, fish, milk, and eggs. The normal body store is 2 to 5 mg in adults, and it usually takes 3 to 4 years to deplete if there is inadequate dietary intake. Vegetarian and vegans often have inadequate dietary intake of cobalamin and may develop low body stores. Other causes of vitamin B-12 deficiency include atrophic gastritis, total or partial gastrectomy, pernicious anemia, and malabsorption in the small bowel secondary to inadequate pancreatic protease and reduction of intrinsic factor-cobalamin receptors in the ileum.
Vitamin K is the cofactor required for gamma-carboxylation of glutamic acid residues on prothrombin, factors VII, IX, X, protein C, and protein S. Vitamin K deficiency occurs as a result of poor nutrition, malabsorption of fat-soluble vitamins, or intrahepatic cholestasis and may be present in any hospitalised or ill patient; patients with inadequate dietary intake are unlikely to develop severe vitamin K deficiency unless concurrently taking antibiotics, when the intestinal source of vitamin K2 is eliminated.
Vitamin C is necessary for the conversion of hydroxyproline to proline, vital for collagen turnover. Inadequate vitamin C intake over a 2- to 3-month period may cause the clinical condition of scurvy manifested by bruising, ecchymoses, and perifollicular hemorrhage.
Clinical signs of chronic liver disease or renal impairment, abdominal masses, splenomegaly, and lymphadenopathy (indicative of bone marrow pathology such as leukemia, lymphoma, or myeloproliferative diseases), bone tenderness (paraproteinemia), and joint swelling (rheumatoid arthritis, connective tissue diseases) may all provide diagnostic information. An assessment should be made of skin elasticity and joint hypermobility, such as found in Ehlers Danlos or pseudoxanthoma elasticum, conditions that may present with microvascular hemorrhage. Infectious causes of purpura include infective endocarditis (clinical signs may include the presence of a new heart murmer, Osler’s nodes, finger clubbing, and splinter hemorrhages) and meningococcal septicemia because of their associated DIC.
Evidence of skin bruising, its site, and extent should be recorded: this includes the presence of petechiae, ecchymoses, and subcutaneous hematoma. The site of purpura may provide diagnostic information: petechiae found on the distal limbs is suggestive of thrombocytopenia, on the face and periorbital area may be suggestive of amyloid and purpura sited on the extensor surfaces of legs, arms, and buttocks is a feature of vasculitic conditions. The presence of spongy-looking bleeding gums, purpura, and corkscrew hairs is suggestive of scurvy. Telangiectasia may appear to look purpuric, but classically these lesions are blanchable. Telangiectasiae affecting the face, mouth, tongue, and lips is diagnostic of hereditary hemorrhagic telangiectasia (HHT), a condition that may cause epistaxis and gastrointestinal bleeding throughout the patient’s life, resulting in severe chronic iron-deficiency anemia. Telangiectasia is also associated with scleroderma, CREST (calcinosis, Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly and telangiectasia) syndrome, and may be evident in patients who have received radiotherapy or solar damage.
Information from the history and physical examination must be carefully correlated with laboratory findings. A patient may be referred for laboratory evaluation of a hemostatic abnormality either as a result of a clinical history of bleeding, or to further assess abnormal blood tests found coincidentally or following routine screening. Screening tests are performed, with a view to focus on more specific tests of platelet, coagulation, and fibrinogen components of the hemostatic system if suggested by historical details, or if any laboratory screening defects are detected (see Table 106-3). No single global assay exists to predict the risk of either bleeding or thrombosis. Several assessments over a period of time may need to be performed to arrive at a diagnosis or an assessment of bleeding risk.
LABORATORY TEST | DETAILS |
|---|---|
Complete blood count | |
Blood film examination | |
Coagulation screen | Prothrombin time (PT) |
Activated partial thromboplastin time (aPTT) | |
Thrombin time | |
Fibrinogen | |
D-dimer or fibrinogen degradation products (FDPs) | |
Tests of thyroid function | TSH, T4, T3 |
Tests of renal function | Urea, creatinine |
Tests of liver function | Albumin, bilirubin, transaminases |
Quantitative Disorders of Platelets
There are numerous causes of isolated thrombocytopenia in the elderly and many are listed in Table 106-4. The most common and relevant causes of thrombocytopenia are discussed here.
Immune | Non-immune |
|---|---|
Immune thrombocytopenia Primary Secondary: SLE Posttransfusion purpura Drug-induced: vancomycin, quinine, heparin | Decreased production Megaloblastic anemia; B-12 and folate deficiency Primary bone marrow disorders: leukemia, lymphoma, myeloma Aplastic anemia Infiltration by solid tumor, TB, sarcoidosis Drugs: chemotherapy Alcohol |
Sequestration: hypersplenism |
Pseudothrombocytopenia is a relatively common phenomenon (found in approximately 0.1% of adults) caused by platelet clumping in the presence of the anticoagulant ethylenediaminetetraacetic acid (EDTA), which is used in vaccutainer tubes for drawing complete blood counts. It should be excluded in any patient with an unexpected low platelet count by examination of the blood film (see Figure 106-2). An accurate platelet count can be estimated by prompt analysis of a fresh blood sample in an alternative anticoagulant such as citrate buffer. Abciximab, a glycoprotein (GP) IIb/IIIa antibody, commonly used in patients with acute coronary syndromes, can cause pseudothrombocytopenia as well as a true drug-induced thrombocytopenia.
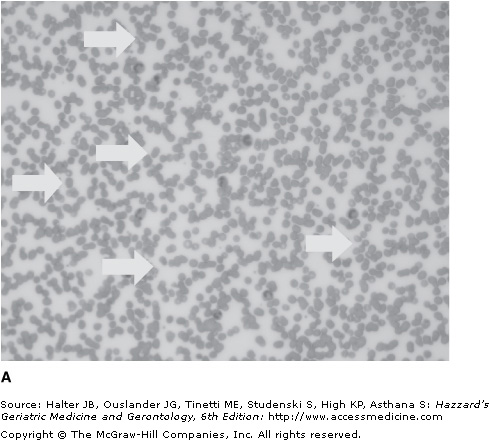
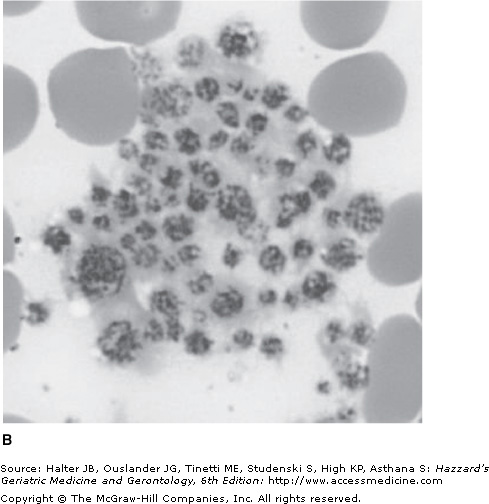
Immune thrombocytopenia (ITP) is an autoimmune disorder characterized by a persistently low platelet count (<150 × 109/L). It is caused by autoantibodies binding to platelet antigen(s) and the resultant premature destruction of the platelets by the reticuloendothelial system, especially the spleen. The diagnosis is made by the exclusion of pseudothrombocytopenia and other causes of true thrombocytopenia. Differentiating between hereditary and acquired thrombocytopenia, especially ITP, is of clinical importance to ensure the patient is not subjected to potentially harmful therapies or procedures (see Table 106-5).
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


