Abstract
This chapter describes the clinical manifestations, diagnosis, complications, and management of sickle cell disease, thalassemia, and other hemoglobinopathies.
Keywords
Sickle cell disease, thalassemia, anemia, transcranial Doppler ultrasonography, transfusion, chelation therapy, iron overload
Sickle Cell Disease
Incidence
Sickle hemoglobin is the most common abnormal hemoglobin found in the United States (approximately 8% of the African-American population has sickle cell trait). The incidence of sickle cell disease (SCD) at birth is approximately one in 600 African-Americans. Fifty percent of the world’s patients with SCD reside in Nigeria, India, and the Democratic Republic of the Congo.
Genetics
- 1.
SCD is transmitted as an autosomal codominant trait.
- 2.
Homozygotes (two abnormal genes, SS) do not synthesize hemoglobin A (HbA); beyond infancy, red cells contain >75% HbS.
- 3.
Compound heterozygotes for HbS and HbC have a form of SCD.
- 4.
Compound heterozygotes for HbS and beta thalassemia trait (beta 0 or beta + ) have a form of SCD (S-beta 0 thalassemia or S-beta+ thalassemia, respectively).
- 5.
Heterozygotes (AS), sickle cell trait, have red cells containing 35–45% HbS.
- 6.
Sickle cell trait provides selective advantage against Plasmodium falciparum malaria (balanced polymorphism).
- 7.
∝-Thalassemia (frequency of one or two ∝ gene deletions is 35% in African-Americans) may be coinherited with sickle cell trait or disease. Individuals who have both ∝-thalassemia and SCD-SS tend to be less anemic than those who have SCD-SS alone. The coinheritance of SCD and ∝-thalassemia trait is associated with a reduction in risk of some complications, such as stroke, but with no effect on the frequency or severity of vaso-occlusive pain.
Pathophysiology
HbS arises as a result of a point mutation (A–T) in the sixth codon of the β-globin gene on chromosome 11, which causes a single amino acid substitution (glutamic acid to valine at position 6 of the β-globin chain). HbS is more positively charged than HbA and hence has a different electrophoretic mobility. Deoxygenated HbS polymerizes, leading to cellular alterations that distort the red cell into a rigid, sickled form. Vaso-occlusion with ischemia–reperfusion injury is the central event, but the underlying pathophysiology is complex, involving a number of factors including hemolysis-associated reduction in nitric oxide bioavailability, chronic inflammation, oxidative stress, altered red cell adhesive properties, activated white blood cells and platelets, altered hemostasis including platelet activation, thrombin activation, lowered levels of anticoagulants, impaired fibrinolysis, and increased viscosity. HbF affects HbS by decreasing polymer content in cells. The effect of HbF on HbS may have direct and indirect effects on other RBC characteristics (i.e., percentage of HbF affects the RBC adhesive properties in patients with SCD). Elevated HbF concentration is associated with a reduction in certain complications of SCD.
Clinical Features
Hematology
- 1.
Anemia—moderate to severe in SS and S-β 0 -thalassemia, milder with SC or Sβ + -thalassemia.
- 2.
Mean corpuscular volume (MCV) is normal with SS; microcytic with concomitant ∝-thalassemia or with S-β-thalassemia.
- 3.
Reticulocytosis.
- 4.
Neutrophilia common.
- 5.
Platelet count often increased.
- 6.
Blood smear—sickle cells (not infants or others with high HbF) increased polychromasia, nucleated red cells, and target cells (Howell–Jolly bodies may indicate hyposplenism).
- 7.
Erythrocyte sedimentation rate—usually low despite inflammation (sickle cells fail to form rouleaux).
- 8.
Hemoglobin electrophoresis—HbS migrates slower than hemoglobin A. Newborn screening shows FS, FSC, or FSA pattern depending on genotype.
Acute Complications
- 1.
Vaso-occlusive pain event ( VOE )
- a.
Episodic microvascular occlusion at one or more sites resulting in pain and inflammation. Common locations and manifestations of VOE are shown in Table 11.1 . Symptoms of fever, erythema, swelling, and focal bone pain may accompany VOE, making it difficult to distinguish from osteomyelitis. Unfortunately, no test clearly distinguishes these two entities but Table 11.2 describes clinical, laboratory, and radiographic features that may be helpful in differentiating bone infarction from osteomyelitis.
Table 11.1
Common Location of Vaso-Occlusive Pain
Site
Manifestations
Hands/feet (dactylitis)
Most common in children younger than 3 years old. Painful swelling of the hands and/or feet. Often can be managed with acetaminophen or nonsteroidal anti-inflammatory medication. Unusual in older children because as the child ages, the sites of hematopoiesis move from peripheral locations such as the fingers and toes to more central locations such as arms, legs, ribs, and sternum
Bone
More common after age 3 years. Often involves long bones, sternum, ribs, spine, and pelvis. May involve more than one site during a single episode. Swelling and erythema may be present. May be difficult to differentiate from osteomyelitis because clinical symptoms, laboratory studies, and radiological imaging may be similar. Features that may aid in distinguishing these two diagnoses are shown in Table 11.2
Abdomen
Caused by microvascular occlusion of mesenteric blood supply and infarction in the liver, spleen, or lymph nodes that results in capsular stretching. Symptoms of abdominal pain and distension mimic acute abdomen
Table 11.2
Differentiation Between Bone Infarction and Osteomyelitis
Features
Favoring osteomyelitis
Favoring vaso-occlusion
History
No previous history
Preceding painful crisis
Pain, tenderness, erythema, swelling
Single site
Multiple sites
Fever
Present
Present
Leukocytosis
Elevated band count (>1000/mm 3 )
Present
Erythrocyte sedimentation rate
Elevated
Normal to low
Magnetic resonance imaging
Abnormal
Abnormal
Bone scan a
Abnormal 99m Tc-diphosphonate
Abnormal 99m Tc-diphosphonate
Normal 99m Tc-colloid marrow uptake
Decreased 99m Tc-colloid marrow uptake
Blood culture
Positive ( Salmonella , Staphylococcus )
Negative
Recovery
Only with appropriate antibiotic therapy
Spontaneous
- b.
The average rate of VOE prompting medical evaluation in SCD-SS is 0.8 events/year. Approximately 40% of patients never seek medical attention for pain, while about 5% of patients account for a third of all VOE. These numbers underestimate the true incidence of VOE because many episodes are managed at home.
- c.
Risk factors for pain include high baseline hemoglobin level, low hemoglobin F levels, nocturnal hypoxemia, and asthma.
- d.
Evidence-based guidelines for the management of VOE are lacking. The typical approach to pain management involves a stepwise progression, beginning with a nonsteroidal anti-inflammatory pain medication for mild to moderate pain and adding an opioid pain medication rapidly if pain is not resolving or for moderate to severe pain. The management of vaso-occlusive pain is shown in Table 11.3 , and a guideline for dosing of commonly utilized pain medications is provided in Table 11.4 . Higher opioid dosing will be required for patients who have developed tolerance. Rapid alleviation of VOE pain is a basic tenet of SCD management.
Table 11.3
Management of Vaso-Occlusive Pain Episodes
At home
Ibuprofen and/or acetaminophen
If continued pain, add oral opiod
Mild pain—codeine
Moderate pain—oxycodone, hydrocodone, morphine
Supportive measures
Heating pad
Fluids
Stool softeners and/or laxative if taking opiods for more than 1–2 days
If pain persists or worsens, patient should be evaluated and treated in an acute care setting
In Emergency Department/Acute Care Unit
Rapid triage and administration of pain medication
If no pain medications were taken prior to arrival and pain not severe, may use ibuprofen and oral opioid
If prior pain medications were taken or pain is severe
Ketorolac tromethamine (NSAID)
IV opioid
Fluids to maintain euvolemia. IV normal saline bolus should only be used if evidence of decreased oral intake/dehydration
Inpatient
Continue nonsteroidal anti-inflammatory agent
Continue IV opioids. Should be given as scheduled medication rather than “as needed”
Consider patient-controlled analgesia pump if pain not adequately controlled
Ongoing evaluation of adequacy of pain control is essential—utilize pain scales
Supportive care
Fluids (oral+IV) to maintain euvolemia
Incentive spirometry
Heating pad—must be used carefully to avoid burns
Bowel regimen to prevent/treat constipation secondary to opioid use
Stool softeners (e.g. docusate)
Laxative (e.g., senna)
Antihistamines (e.g., diphenhydramine, hydroxyzine) for pruritis
Transition to oral nonsteroidal and oral opiod as pain level improves. Addition of long-acting opioid (e.g., sustained-release morphine)
Table 11.4
Dosages of Common Analgesics for Management of Sickle Cell Vaso-Occlusive Pain
Medication
Usual dose
Maximum dose
Route
Interval (h)
NONSTEROIDAL ANTI-INFLAMMATORY MEDICATIONS
Ibuprofen
10 mg/kg
800 mg
PO
Q 6–8
Naproxen
5–7 mg/kg
500 mg
PO
Q 12
Ketorolac
0.5 mg/kg
30 mg
IV, IM
Q 6–8 a
OPIOID PAIN MEDICATIONS
Codeine
0.5–1 mg/kg
60 mg
PO
Q 4–6
Oxycodone
<6 years
0.05–0.15 mg/kg
2.5 mg
PO
Q 4–6
6–12 years
0.05–0.2 mg/kg
5 mg
PO
Q 4–6
>12 years
0.05–0.2 mg/kg
10 mg
PO
Q 4–6
Hydromorphone
<50 kg
0.03–0.08 mg/kg
PO
Q 3–4
≥50 kg
1–4 mg/dose
8 mg
PO
Q 3–4
<50 kg
0.01 mg/kg
IV, IM, SQ
Q 3–4
≥50 kg
0.01 mg/kg
4 mg
IV, IM, SQ
Q 3–4
Morphine (immediate release)
<6 months
0.1–0.3 mg/kg
PO
Q 3–4
6 months-18 years
0.2–0.5 mg/kg
PO
Q 3–4
Adults
10–30 mg/dose
PO
Q 3–4
Morphine (controlled release)
>30 kg
0.3–0.6 mg/kg
60 mg
PO
Q 8–12
Morphine
<6 months
0.05–0.1 mg/kg
IV, IM, SQ
Q 3–4
≥6 months
0.1–0.2 mg/kg
15 mg
IV, IM, SQ
Q 3–4
PO, orally; IV, intravenously; IM, intramuscularly; SQ, subcutaneously.
- e.
Other drug therapies are under investigation for treatment of acute VOE, but are not yet clinically available. A number of treatments have been tested or are currently being tested in early Phase 1 or 2 trials for patients with SCD and acute VOE. These include:
- i.
Drugs and supplements that cause vasodilatation, including arginine and nitrous oxide
- ii.
Drugs that inhibit platelet aggregation
- iii.
Drugs that lower blood viscosity
- iv.
Drugs that affect red blood cell adhesion
- i.
- f.
Prevention of pain
- i.
Hydroxyurea (HU)
- ii.
Prophylactic red blood cell transfusions
- i.
- a.
- 2.
Acute chest syndrome ( ACS )
- a.
ACS is the most common cause of death and the second most common cause of hospitalization in children with SCD. It is generally defined as the development of a new pulmonary infiltrate accompanied by symptoms including fever, chest pain, tachypnea, cough, hypoxemia, and wheezing.
- b.
ACS is caused by infection, infarction, and/or fat embolization and iatrogenically by overhydration. About 50% of ACS events are associated with infections, including viruses, atypical bacteria including Mycoplasma and Chlamydia , and less frequently with Streptococcus pneumoniae . Parvovirus B19 infection can also result in ACS. In about half of cases, ACS develops during hospitalization, often for vaso-occlusive pain, where fat embolization, hypoventilation, and iatrogenic overhydration contribute to the pathophysiology.
- c.
The incidence of ACS in SCD-SS is about 24 events per 100 patients in children. The incidence in other sickle cell genotypes is lower (SS>Sβ 0 -thalassemia>SC>Sβ + -thalassemia), and concomitant α-thalassemia does not appear to affect ACS rates.
- d.
The risk of ACS is directly proportional to the hemoglobin level and white blood cell count; increased levels of cytokines and/or white cell adhesion to the endothelium may play a role. Rates of ACS are also higher in children with asthma. Higher hemoglobin F levels appear to be protective.
- e.
Laboratory findings:
- i.
White blood cell count is often elevated.
- ii.
Hemoglobin level often falls to 1.5 g/dl below baseline values.
- iii.
Thrombocytosis may be present, and often follows an episode of ACS.
- i.
- f.
The management of ACS is described in Table 11.5 .
Table 11.5
Management of Acute Chest Syndrome in Children
Evaluations
Chest radiograph
Complete blood count and reticulocyte count
Blood type and screen
Blood culture
Viral studies
Pulse oximetry
Consider arterial blood gas in room air
Treatment
Antibiotics: Broad-spectrum intravenous antibiotic such as cefuroxime plus an oral macrolide (erythromycin or azithromycin) to cover atypical bacteria
Supplemental oxygen if hypoxemic
Fluids: Intravenous+oral fluids should be kept at maintenance. Avoid overhydration.
Pain control: Must be carefully monitored. Goal is to relieve pain to reduce splinting/poor aeration but avoid oversedation with hypoventilation
Transfusion:
Simple transfusion (10–15 cc/kg)—do not exceed post transfusion hemoglobin level of ~10 g/dl
Exchange transfusion—if no improvement with simple transfusion or with severe hypoxemia/respiratory distress
Bronchodilators—particularly if history of reactive airways disease or if wheezing present
Steroids may be beneficial for severe acute chest syndrome or if reactive airways disease component. There is a risk of rebound pain with discontinuation of the steroids
Incentive spirometry to reduce atelectasis
Mechanical ventilation as needed
Consider thoracentesis if significant pleural effusion
- g.
Prevention of ACS:
Patients with a history of recurrent ACS are candidates for preventative/curative therapies including;
- i.
Hydroxyurea (HU).
- ii.
Prophylactic red cell transfusions. Optimal target HbS level is not known, but usually a goal of 30–50% is used.
- iii.
Stem cell transplantation.
- i.
- a.
- 3.
Overt stroke
- a.
Acute symptomatic stroke is usually infarctive in children, although hemorrhagic stroke may occur, particularly in older children and adults.
- b.
The most common underlying lesion is intracranial arterial stenosis or occlusion, usually involving the large arteries of the circle of Willis, particularly the distal internal carotid artery (ICA) and the middle (MCA) and anterior cerebral arteries (ACAs).
- c.
Chronic injury to the endothelium of vessels by sickled red blood cells results in changes in the intima with proliferation of fibroblasts and smooth muscle; the lumen is narrowed or completely obliterated. Small friable collateral blood vessels known as moyamoya may develop. Infarction of brain tissue occurs acutely as a result of in situ occlusion of the damaged vessel or distal embolization of a thrombus. Perfusional and/or oxygen delivery deficits related to changes in blood pressure or other factors also may contribute to infarction, particularly in watershed zones.
- d.
Stroke is most common in homozygous SCD-SS. Prior to transcranial Doppler (TCD) ultrasound screening with transfusions for high-risk children, stroke prevalence in children with SCD-SS was estimated at 11%, with the highest incidence rates occurring in the first decade of life (1.02 per 100 patient-years in 2–5 year olds and 0.79 per 100 patient-years in 6–9 year olds).
- e.
A number of clinical, laboratory, and radiological risk factors for stroke have been identified ( Table 11.6 ).
Table 11.6
Factors Associated with Increased Risk of Overt Infarctive Stroke in Sickle Cell Disease
Clinical
History of transient ischemic attacks
History of bacterial meningitis
Sibling with SCD-SS and stroke
Recent episode of acute chest syndrome (within 2 weeks)
Frequent acute chest syndrome
Systolic hypertension
Nocturnal hypoxemia
Laboratory
Low steady-state hemoglobin level
No alpha gene deletion
Certain HLA haplotypes
Radiological
Abnormal transcranial Doppler ultrasound
Silent infarct
- f.
Symptoms of stroke include:
- i.
Focal motor deficits (e.g., hemiparesis, gait dysfunction).
- ii.
Speech defects.
- iii.
Altered mental status.
- iv.
Seizures.
- v.
Headache.
- i.
- g.
Gross neurological recovery occurs in approximately two-thirds of children, but neurocognitive deficits are common.
- h.
In untreated patients, about 70% of patients experience a recurrence within 3 years. Outcome after recurrent stroke is worse.
- i.
Any child with SCD who develops acute neurological symptoms requires immediate medical evaluation. A guideline for management is presented in Figure 11.1 . The acute management involves prompt diagnosis and treatment.
- •
Diagnosis :
- •
Physical examination with detailed neurological examination. Treatment in the face of clinical suspicion must not await imaging confirmation. Head CT scan is useful for detecting intracranial hemorrhage and often more readily available than magnetic resonance imaging (MRI). May not be positive for acute infarction within the first 6 h.
- •
Brain MRI with diffusion-weighted imaging is more sensitive to early ischemic changes and may be abnormal within 1 h. Should be performed as soon as possible in a child with SCD presenting with acute neurological symptoms, but should not delay empiric treatment.
- •
Magnetic resonance arterial angiography (MRA)—demonstrates large-vessel disease.
- •
- •
Treatment :
- •
Transfusion. Exchange transfusion, either automated or manual, should be performed as soon as possible. The goal is to reduce the amount of HbS to less than 30% and to raise the hemoglobin level to approximately 10 g/dl. If exchange transfusion is not readily available, a simple transfusion to raise the hemoglobin level to no greater than 10 g/dl, although suboptimal, may be used. Exchange transfusion may be associated with a decreased risk of stroke recurrence.
- •
Supportive therapy including avoiding hypotension and maintaining adequate oxygenation and euthermia should be initiated as adjunctive therapy.
- •
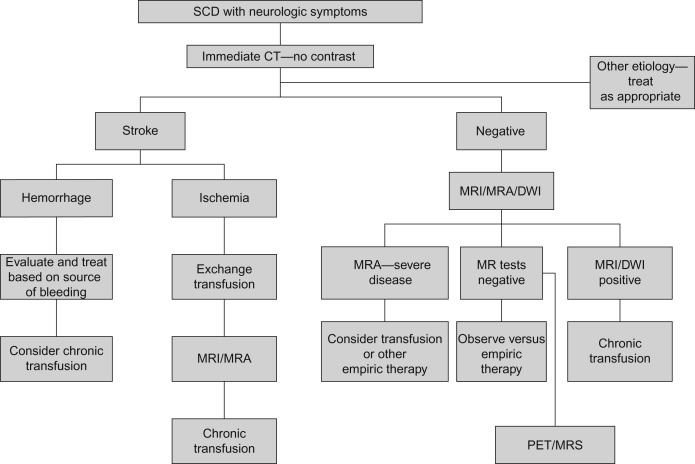
Figure 11.1
Management of the child with sickle cell disease and neurological symptoms. PET, positron emission tomography; MRA, magnetic resonance arterial angiography; DWI, diffusion-weighted imaging; MRS, magnetic resonance spectroscopy.
From The Management of Sickle Cell Disease, 4th Ed, 2002, National Heart, Lung and Blood Institute of the National Institutes of Health and The US Department of Health and Human Services, NIH Publication No. 02-2117, with permission.
- •
- j.
Long-term management
- i.
Prevention of recurrent stroke
- •
A chronic red cell transfusion program should be instituted, with the goal of maintaining the pretransfusion HbS level at less than 30%. Transfusions must be continued indefinitely, due to the high risk of stroke recurrence after discontinuation of therapy. After a period of 3–4 years after the initial stroke, it may be possible to allow the pretransfusion HbS level to rise to less than 50% in low-risk patients, without increased risk of stroke recurrence. This approach is associated with decreased transfusional iron loading. The high erythroid drive in SCD permits a transfusion approach in which chronic exchange transfusion to a hemoglobin that does not suppress erythropoiesis allows for erythropoiesis that utilizes iron and still permits a HbS<30 resulting in iron balance rather than iron loading.
- •
Hematopoietic stem cell transplantation (HSCT).
- •
The uses of revascularization procedures such as encephalodurosynangiosis are controversial and believed by some to be beneficial in children with significant vasculopathy, particularly if symptomatic (transient ischemic attacks, recurrent stroke) although published data on use in SCD are limited.
- •
Treatment with HU with a several month overlap period with transfusions (until the hematological effects of HU were evident), was associated with a stroke recurrence rate of 3.6 events per 100 patient-years, which is lower than the risk of stroke recurrence without treatment. Furthermore, the use of phlebotomy during treatment with HU successfully reduced iron stores. However, multicenter phase 3 trial of HU and phlebotomy compared with transfusions and chelation therapy was terminated due to a higher rate of recurrent stroke (10%) in the HU arm compared with continued transfusions (0%) without any difference in iron reduction between the two treatment arms.
- •
Prophylactic aspirin may also be useful in children with progressive vasculopathy, but the risks of hemorrhage must be weighed against the potential benefit.
- •
- ii.
Rehabilitation
- •
Physical and occupational therapy as needed.
- •
Neuropsychological testing should be performed with educational interventions if indicated.
- •
- i.
- k.
Primary stroke prevention
- i.
Screening
- •
TCD ultrasonography is a noninvasive study used to measure the blood flow velocity in the large intracranial vessels of the circle of Willis.
- •
The highest time-averaged mean velocity (TAMMvel) in the ICA, its bifurcation, and the MCA are used to categorize studies into risk groups:
- •
Normal (velocity <170 cm/sec), low risk.
- •
Conditional (170–199 cm/sec), moderate risk.
- •
Abnormal (≥200 cm/sec), high risk.
- •
Inadequate—unable to obtain velocity in the ICA or MCA on either side, in the absence of a clearly abnormal value in another vessel. Inadequate TCD may be due to technique, skull thickness, or severely stenosed vessel.
- •
- •
Very low velocity (ICA/MCA velocity <70 cm/s) may indicate vessel stenosis and increased risk of stroke.
- •
Elevated velocity in the ACA is associated with increased stroke risk. Treatment of children with isolated high ACA velocities has not been established. Brain MRI/A should be obtained. Chronic transfusion should be instituted for children with ACA velocity ≥200 cm/s, and any child if silent infarcts and/or cerebral blood vessel stenosis are present on MRI/A.
- •
TCD screening is recommended for children with SCD-SS or SCD-Sβ 0 -thalassemia ages 2–16 years. Screening is performed annually, but more frequently if the prior study was not normal. An approach to screening is shown in Table 11.7 . In addition, more frequent screening should also be considered if other known stroke risk factors are present (such as sibling with SCD-SS and stroke or abnormal TCD).
Table 11.7
Transcranial Doppler Ultrasonography Screening Protocol
Last TCD result (TAMMvel in ICA/MCA)
Screening interval
Normal (<170 cm/s)
Annual
Low conditional (170–184 cm/s)
3–6 months a
High conditional (185–199 cm/s)
6 weeks–3 months a
Abnormal (200–219 cm/s)
1 week
High abnormal (220 cm/s or higher)
No confirmation needed—recommend treatment
- •
Brain MRI/MRA should be obtained in children with abnormal TCD and should be considered for children with conditional TCD.
- •
Brain MRA is helpful to evaluate cerebral vasculature in children with repeatedly inadequate TCD or with very low velocity.
- •
- ii.
Treatment
- •
Chronic transfusion to maintain the HbS level <30% reduces the risk of stroke by >90% in children with abnormal TCD.
- •
Discontinuation of transfusion therapy after at least 30 months of transfusion with normalization of TCD results is associated with a high risk of reversion to abnormal TCD and stroke. Thus, transfusions are continued indefinitely.
- •
Stem cell transplantation with an HLA-identical sibling donor should be considered.
- •
HU therapy is associated with a lowering of TCD velocities and is currently under study for primary stroke prevention.
- •
- i.
- a.
- 4.
Priapism
- a.
Priapism is a sustained, painful erection of the penis. Priapism may be prolonged (lasts more than 3 h), or stuttering (lasts less than 3 h). Stuttering episodes often recur or may develop into a prolonged episode.
- b.
Occurs in 30–45% of patients with SCD, most commonly in the SS type. The prevalence is likely underestimated due to underreporting by patients.
- c.
Mean age at the first episode of priapism in patients with SCD is about 12–15 years; 75% have their first episode before age 20 years.
- d.
Priapism often occurs in the early morning hours, when normal erections occur, and is probably related to nocturnal acidosis and dehydration. The normal slow blood flow pattern in the penis is similar to the blood flow in the spleen and renal medulla. Failure of detumescence is due to venous outflow obstruction or to prolonged smooth muscle relaxation, either singly or in combination.
- e.
A history of priapism in childhood is associated with later sexual dysfunction, with 10–50% of adults with SCD and a history of priapism reporting impotence.
- f.
Treatment
- i.
At home, patients may try warm baths, oral analgesics, increased oral hydration, and pseudoephedrine.
- ii.
Patients should be evaluated in an emergency room for episodes lasting over 2 h.
- iii.
Initial treatment includes intravenous hydration and parenteral analgesia.
- iv.
Episodes lasting ≥4 h are associated with an increased risk of irreversible ischemic injury and thus warrant more aggressive management. Urological consultation should be obtained. Treatment involves aspiration of the corpus cavernosum followed by irrigation with or without intracavernous administration of a dilute (1:1,000,000) epinephrine solution. Although published data in SCD are lacking, a dilute solution of phenylephrine, an alpha adrenergic agent, rather than epinephrine, has also been utilized in some centers.
- v.
The role of transfusion for the management of priapism is controversial and the clinical response is variable. Furthermore, exchange transfusion for acute priapism has been associated with the development of acute neurological events.
- vi.
Surgical shunting procedures (cavernosa spongiosum or cavernosaphenous vein) may be considered if the above treatments fail although shunt occlusion is a common complication.
- i.
- g.
Prevention of priapism
- i.
Pseudoephedrine, 30–60 mg orally at bedtime.
- ii.
HU therapy has been employed, although this treatment has not been studied for this indication.
- iii.
Leuprolide injections, a gonadotropin-releasing hormone analog that suppresses the hypothalamic–pituitary access, reducing testosterone production.
- iv.
Phosphodiesterase type 5 inhibitors (excluding Sildenafil) may have some benefit, though further research is needed.
- v.
Transfusion protocol for 6–12 months following an episode of priapism requiring irrigation and injection.
- i.
- a.
- 5.
Splenic sequestration
- a.
Highest prevalence between 5 and 24 months of age in SCD-SS (may occur at older ages in other sickling syndromes).
- b.
May occur in association with fever or infection.
- c.
Splenomegaly due to pooling of large amounts of blood in the spleen.
- d.
Rapid onset of pallor and fatigue. Abdominal pain is often present.
- e.
Hemoglobin level may drop precipitously, followed by hypovolemic shock and death.
- f.
Reticulocytosis and nucleated red blood cells often present.
- g.
Platelet and white blood cell count also usually fall from baseline.
- h.
Treatment of splenic sequestration is shown in Table 11.8
Table 11.8
Management of Splenic Sequestration
Treatment of acute splenic sequestration episode
Monitor cardiovascular status, spleen size, and hemoglobin level closely
Normal saline bolus of 10–20 cc/kg
Red cell transfusion. Administer in small aliquots because transfusion often results in reduction in spleen size with “autotransfusion” of previously trapped red cells. Rapid infusion used for cardiovascular instability
Pain management
Prevention of recurrent splenic sequestration
Splenectomy if history of one major or two minor acute splenic sequestration episodes
For children <2 years old, chronic transfusion therapy can be considered to postpone splenectomy
- a.
- 6.
Transient pure red cell aplasia
- a.
Cessation of red cell production that may persist for 7–14 days with profound drop in hemoglobin (as low as 1 g/dl).
- b.
Reticulocyte count and the number of nucleated red cells in the marrow sharply decrease; platelet and white blood cell counts are generally unaffected.
- c.
May occur in several members of a family and can occur at any age.
- d.
Almost invariably associated with parvovirus B19 infection.
- e.
Terminates spontaneously usually after about 10 days (recovery occurs with reticulocytosis and nucleated red cells in the blood).
- f.
Vaso-occlusive pain and/or splenic sequestration may occur in association with parvovirus B19/transient pure red cell aplasia.
- g.
Treatment
- i.
Close monitoring of complete blood count (CBC) and reticulocyte count.
- ii.
Red cell transfusion to raise hemoglobin level to no greater than 9–10 g/dl.
- iii.
Monitor siblings with SCD closely (CBC, reticulocyte count, parvovirus polymerase chain reaction (PCR), and/or titers).
- i.
- a.
Chronic Complications and End-Organ Damage
- 1.
Central nervous system
- a.
Silent stroke
- i.
Defined as one or more focal T2-weighted signal hyperintensities demonstrated on brain MRI, in the absence of a focal neurological deficit corresponding to the anatomical distribution of the brain lesion.
- ii.
Present in approximately 35% of children with SCD-SS and occurs less commonly in other sickle cell genotypes.
- iii.
Associated with neuropsychologic deficits and impaired school performance.
- iv.
Silent infarcts may progress in size and number over time and are associated with an increased risk of overt stroke.
- a.
Treatment
- i.
Management of children with silent infarcts includes neuropsychological testing and monitoring of academic performance.
- ii.
Chronic transfusion therapy to maintain the hemoglobin level above 9 g/dl and the HbS below 30% for children with silent cerebral infarcts is associated with a reduction in infarct recurrence. New or enlarged silent infarcts or overt stroke occurred in 6% of children receiving transfusions compared with 14% of children in the observation group.
- iii.
HU has not been studied for this indication.
- i.
- i.
- 2.
Cardiovascular system
- a.
Abnormal cardiac findings are present in most patients as a result of chronic anemia and the compensatory increased cardiac output.
- b.
Cardiomegaly is found in most patients and left ventricular hypertrophy occurs in about 50%.
- c.
Prolonged QTc >440 msec occurs in 9–38% of children with SCD, most commonly the SS type. Prolonged QTc is associated with increased risk of mortality in adults with SCD.
- d.
A moderate-intensity systolic flow murmur is often present.
- e.
Echocardiogram may show left and right ventricular dilatation, increased stroke volume, and abnormal septal motion.
- f.
Pulmonary hypertension
- i.
Defined as a resting pulmonary artery systolic pressure equal to or greater than 25 mmHg. Right heart catheterization is required to make a definitive diagnosis. Noninvasive echocardiography often is used to screen for the possible presence of pulmonary hypertension. Tricuspid regurgitant jet velocity (TRV) of at least 2.5 m/s is an indicator of possible pulmonary hypertension.
- ii.
Prevalence of pulmonary hypertension documented by right heart catheterization in adults is estimated at 6–11%, with 10% of these adults having moderate to severe pulmonary hypertension (pressure above 45 mmHg). An elevated TRV is found in approximately 30% of adults with SCD. The prevalence of pulmonary hypertension in children appears to be about 11% and is most common with the SS genotype. Diagnosis of pulmonary hypertension by TRV alone has been questioned. Children with elevated TRV should be managed along with a cardiologist.
- iii.
In adults, pulmonary hypertension by right heart catheterization, elevated TRV, and increased serum N-terminal pro-brain natriuretic peptide are independent risk factors for mortality; the significance of these findings in children is unclear.
- iv.
A central role for hemolysis and altered NO bioavailability has been postulated.
- v.
The optimal treatment is unknown, but HU or red cell transfusions have been used. Treatment with sildenafil, an agent used to treat pulmonary hypertension in other patient groups, is associated with an increased risk of vaso-occlusive pain episodes.
- i.
- a.
- 3.
Pulmonary
- a.
Reduced PaO 2 .
- b.
Reduced PaO 2 saturation. Pulse oximetry may not correlate with PaO 2 in steady state. Changes in pulse oximetry are useful for monitoring children with ACS. Daytime and/or nocturnal hypoxemia may be present.
- c.
Pulmonary fibrosis—Chronic lung disease: Early identification of progressive lung disease using pulmonary function testing is imperative. Aggressive treatment has little benefit in end-stage lung disease and this should be avoided by prophylactic transfusions.
- d.
Asthma—Prevalence appears to be higher than in the general population in children with SCD. Asthma is associated with complications of SCD including pain, ACS, stroke, and pulmonary hypertension. Aggressive management is warranted.
- a.
- 4.
Kidney
- a.
Increased renal flow.
- b.
Increased glomerular filtration rate.
- c.
Enlargement of kidneys; distortion of collecting system.
- d.
Hyposthenuria (urine concentration defect): Hyposthenuria is the first manifestation of sickle cell-induced obliteration of the vasa recta of the renal medulla. Edema in the medullary vasculature is followed by focal scarring, interstitial fibrosis, and destruction of the countercurrent mechanism. Hyposthenuria results in a concentration capacity of more than 400–450 mOsmol/kg and an obligatory urinary output as high as 2000 ml/m 2 /day, causing the patient to be particularly susceptible to dehydration. The increased urine output is associated with nocturia, often manifesting as enuresis.
Treatment of nocturnal enuresis includes behavioral modifications such as waking to void and the use of a bedwetting alarm. 1-deamino-8-D-arginine vasopressin at bedtime has also been used. The intranasal form is no longer recommended for childhood nocturnal enuresis. An oral dose of 0.2 mg has been used in children over age 6 years.
- e.
Hematuria: Papillary necrosis is usually the underlying anatomic defect. Treatment of papillary necrosis is IV hydration and rest. Frank hematuria usually resolves, although bleeding can be prolonged. Antifibrinolytic agents such as epsilon-aminocaproic acid have been used for recalcitrant bleeding with variable success. However, great caution must be taken when using this drug because of the risk of thrombosis and urinary obstruction. Evaluation for other causes of hematuria (e.g., renal medullary carcinoma) is indicated for the first episode of hematuria.
- f.
Renal tubular acidification defect.
- g.
Increased urinary sodium loss (may result in hyponatremia).
- h.
Hyporeninemic hypoaldosteronism and impaired potassium excretion are results of renal vasodilating prostaglandin increase in patients with SCD.
- i.
Proteinuria: Persistent increasing proteinuria is an indication of glomerular insufficiency, perihilar focal segmental sclerosis, and renal failure. Intraglomerular hypertension with sustained elevations of pressure and flow is the prime etiology of the hemodynamic changes and subsequent proteinuria. If proteinuria persists for more than 4–8 weeks, angiotensin-converting enzyme inhibitors (i.e., enalapril) are recommended.
- j.
Nephrotic syndrome: A 24-h urine protein of more than 2 g/day, edema, hypoalbuminemia, and hyperlipidemia may indicate progressive renal insufficiency. The efficacy of steroid therapy in the management of nephrotic syndrome in SCD is not clear. Carefully monitored use of diuretics is indicated to control edema.
- k.
Chronic renal failure: Uremia. Renal failure can be managed with peritoneal dialysis, hemodialysis, and transplantation.
- a.
- 5.
Liver and biliary system
- a.
Chronic hepatomegaly.
- b.
Liver function tests: Increased serum aspartate transaminase and serum alanine transaminase.
- c.
Cholelithiasis
- i.
Chronic hemolysis with increased bilirubin turnover causes pigmented stones.
- ii.
Occurs as early as 2 years old and affects at least 30% by age 18 years.
- iii.
Sonographic examinations of the gallbladder should be performed in children with symptoms.
- iv.
The treatment for symptomatic cholelithiasis is laparoscopic cholecystectomy. The role of screening and treatment of asymptomatic patients is unclear.
- i.
- d.
Transfusion-related hepatitis. Hepatitis C is more common in older patients who received red cell transfusions prior to the availability of screening of blood products.
- e.
Intrahepatic crisis: Intrahepatic sickling can result in massive hyperbilirubinemia, elevated liver enzyme values, and a painful syndrome mimicking acute cholecystitis or viral hepatitis. Progression to multiorgan system failure may occur. Early exchange transfusion is indicated.
- f.
Hepatic necrosis, portal fibrosis, regenerative nodules, and cirrhosis are common postmortem findings that may be a consequence of recurrent vascular obstruction and repair.
- g.
Transfusional iron overload, secondary to repeated intermittent or chronic transfusions may cause hepatic fibrosis.
- a.
- 6.
Bones
Skeletal changes in SCD are common because of expansion of the marrow cavity, bone infarcts, or both.
- a.
Avascular necrosis (AVN): The most common cause of AVN of the femoral head is SCD. The incidence is much higher with coexistent α-thalassemia in patients who have frequent painful events and in those with the highest hematocrits. The pathophysiology is sludging in marrow sinusoids, marrow necrosis, healing with increased intramedullary pressure, bone resorption, and eventually collapse. About 50% of patients are asymptomatic. Symptomatic patients have significant chronic pain and limited joint mobility. The diagnosis is made radiographically and shows:
- i.
Subepiphyseal lucency and widened joint space.
- ii.
Flattening or fragmentation and scarring of the epiphysis.
- iii.
On MRI, AVN of femoral head can be detected before deformities are apparent on radiograph.
- •
Treatment:
Therapy for AVN is largely supportive, with bed rest, nonsteroidal anti-inflammatory drugs (NSAIDs), and limitation of movement during the acute painful episode. Transfusion therapy and HU do not seem to delay progression of AVN. Physical therapy is helpful and may reduce the risk of progression. Core decompression of the affected hip has been reported to reduce pain and stop progression of the disease. In this procedure, avascularized bone is removed to decompress the area with the potential for subsequent new bone formation. This procedure seems to be beneficial only in the early stages of AVN and before loss of the integrity of the femoral head. AVN of the hip may have its onset in childhood, so thorough musculoskeletal examination with concentration on the hips should be performed at least yearly in children with SCD. This ensures that AVN is detected early when it is in its most treatable form. Total hip replacement may be the only option for severely compromised patients; 30% of replaced hips require surgical revision within 4.5 years, and more than 60% of patients continue to have pain and limited mobility postoperatively. AVN of the humeral head is less common. Patients are less symptomatic, and arthroplasty is exceedingly rare.
- •
- i.
- b.
Widening of medullary cavity and cortical thinning: Hair-on-end appearance of skull on radiograph.
- c.
Fish-mouth vertebra sign on radiograph.
- a.
- 7.
Eyes
- a.
Retinopathy: Sickle retinopathy is common in all forms of SCD, but particularly in those patients with SCD, type SC.
- i.
Nonproliferative: Occlusion of small blood vessels of the eye detected on dilated ophthalmological exam and usually not associated with defects in visual acuity.
- ii.
Proliferative: Occlusion of small blood vessels in the peripheral retina may be followed by enlargement of existing capillaries or development of new vessels. Clusters of neovascular tissue “sea fans” grow into vitreous and along the surface of the retina. Sea fans may cause vitreous hemorrhage, which results in transient or prolonged loss of vision. Small hemorrhages resorb, but repeated leaks cause formation of fibrous strands. Shrinkage of these strands can cause retinal detachment.
- •
Treatment:
- •
Nonproliferative: Treatment not usually needed.
- •
Proliferative: Neovascularization may not progress or may even regress spontaneously.
Indications for treatment include bilateral proliferative disease, rapid growth of neovascularization, and large elevated neovascular fronds. Laser photocoagulation and other methods are used to induce regression of neovascularization. With proper screening and new methods such as laser surgery, most of the complications of retinopathy can be avoided. Annual ophthalmologic examinations including inspection of the retina are indicated for children from the age of 5 years for children with SCD-SC and 8 years for children with SCD-SS.
- •
- •
- i.
- b.
Angioid streaks: These are pigmented striae in the fundus caused by abnormalities in the Baruch membrane due to iron or calcium deposits or both. They usually produce no problems for the patient, but occasionally they can lead to neovascularization that can bleed into the macula and decrease vision.
- c.
Hyphema: Blood in the anterior chamber (hyphema) rarely occurs secondary to sickling in the aqueous humor, because of its low pH and pO 2 . Traumatic hyphema may occur as in any individual. Anterior chamber paracentesis should be performed if pressure is increased.
- d.
Conjunctivae: Comma-shaped blood vessels, seemingly disconnected from other vasculature, can be seen in the bulbar conjunctiva of patients with SCD and variants (SS>SC>Sβ-thalassemia). These produce no clinical disability. Their frequency may be related to the number of irreversibly sickled cells in the blood. This abnormality can be identified by using the +40 lens of an ophthalmoscope.
- a.
- 8.
Ears
Up to 12% of patients have high-frequency sensorineural hearing loss. The pathophysiology may involve sickling in the cochlear vasculature with destruction of hair cells.
- 9.
Adenotonsillar hypertrophy
Adenotonsillar hypertrophy giving rise to upper airway obstruction can become a problem from the age of 18 months. The marked hypertrophy is postulated to be compensation for the loss of lymphoid tissue in the spleen. It occurs in at least 18% of patients. In severe cases, this can cause hypoxemia at night with consequent sickling. Early tonsillectomy and adenoidectomy may be indicated in these patients.
- 10.
Skin
Cutaneous ulcers of the legs occur over the external or internal malleoli. Leg ulcers occur less commonly in children, and rarely before age 10 years. Ulcers are most common in homozygous SCD. Ulceration may result from increased venous pressure in the legs caused by the expanded blood volume in the hypertrophied bone marrow.
Treatment:
- a.
Rest; elevation of the leg.
- b.
Protection of the ulcer by the application of a soft sponge–rubber doughnut.
- c.
Debridement and scrupulous hygiene.
- d.
Low-pressure elastic bandage and above-the-knee elastic stockings to improve venous circulation.
- e.
Transfusion therapy for 3–6 months course if ulcers persist despite optimal care.
- f.
Antibiotic therapy if acutely infected (typical organisms are Staphylococcus , Streptococcus , and Pseudomonas species).
- g.
Oral administration of zinc sulfate (220 mg three times a day) may promote healing of leg ulcers.
- h.
Split-thickness skin grafts.
- a.
- 11.
Growth and development
- a.
Birth weight is normal. However, by 2–6 years of age, the height and weight are significantly delayed. The weight is more affected than the height, and patients with SCD-SS and Sβ 0 -thalassemia experience more delay in growth than patients with SCD-SC and Sβ + -thalassemia. In general, by the end of adolescence, patients with SCD have caught up with controls in height but not weight. The poor weight gain is likely to represent increased caloric requirements in anemic patients with increased bone marrow activity and cardiovascular compensation. Zinc deficiency may be a cause of poor growth. In these patients, zinc supplementation (dose of 220 mg three times a day) at about 10 years of age should be administered. Growth hormone levels and growth hormone stimulation studies appear to be normal in most children who have impaired growth.
- b.
Delayed sexual maturation: Tanner 5 is not achieved until the median ages of 17.3 and 17.6 years for girls and boys, respectively. In males, decreased fertility with abnormal sperm motility, morphology, and numbers is prominent. Zinc sulfate 220 mg three times a day may be effective for sexual maturity in these patients; females are more responsive than males.
- a.
- 12.
Functional hyposplenism
- a.
By 6 months of age, mild splenomegaly may be apparent and persists during early childhood, after which the spleen undergoes progressive fibrosis (autosplenectomy).
- b.
Functional reduction of splenic activity occurs in early life. This is the consequence of altered intrasplenic circulation caused by intrasplenic sickling. It can be temporarily reversed by transfusion of normal red cells. Children with functional hyposplenia are 300–600 times more likely to develop overwhelming pneumococcal and Haemophilus influenzae sepsis and meningitis than are normal children; other organisms involved are Gram-negative enteric organisms and Salmonella . The period of greatest risk of death from severe infection occurs during the first 5 years of life.
- c.
Functional hyposplenism may be demonstrated by the following:
- i.
Presence of Howell–Jolly bodies on blood smear.
- ii.
99m Tc-gelatin sulfur colloid spleen scan—no uptake of the radioactive colloid by enlarged spleen.
- iii.
Pitted red blood cell count >3.5%.
- i.
- a.
Diagnosis
- 1.
In utero: SCD can be diagnosed accurately in utero by mutation analysis of DNA prepared from chorionic villus biopsy or fetal fibroblasts (obtained by amniocentesis). With the advent of PCR amplification of specific DNA sequences, sufficient DNA can be obtained from a very small number of fetal cells, thereby eliminating the necessity of culturing fetal fibroblasts from amniotic fluid. These techniques should be employed before 10 weeks’ gestation.
- 2.
During the newborn period: The diagnosis of SCD can be established by electrophoresis using:
- a.
Isoelectric focusing (most commonly used in screening programs).
- b.
High-performance liquid chromatography.
- c.
Citrate agar with a pH of 6.2, a system that provides distinct separation of HbS, HbA, and HbF.
- d.
DNA-based mutation analysis.
These tests are commonly performed on a dried blood specimen blotted on filter paper (Guthrie cards) used in newborn screening programs.
- a.
- 3.
In older children: Table 11.9 lists the diagnosis and differential diagnosis of various sickle cell syndromes.
Table 11.9Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree