Fig. 1.1
Site of hematopoiesis during fetal development. Hematopoietic stem cells (HSCs) are thought to derive from the ventral mesoderm. The initial blood production termed as “primitive hematopoiesis” occurs in the yolk sac, producing red blood cells to supply oxygen for rapidly growing tissue. This primitive hematopoiesis is rapidly replaced by adult-type hematopoiesis termed as “definitive hematopoiesis,” which produces HSCs that have the ability to give rise to all hematopoietic lineages. Definitive hematopoiesis, occurring in aorta-gonad-mesonephros (AGM) region, is also found in the placenta, where it moves from the fetal liver to the bone marrow to generate hematopoietic cells throughout life
1.2.2 Identification of HSCs
HSCs demonstrate long-lasting self-renewal ability and differentiate into all types of hematopoietic cells. Self-renewal is the ability of a cell to produce itself by cell division, whether symmetrical or asymmetrical, and this self-renewal ability is the essential characteristic of all stem cells, including HSCs.
HSCs are an extremely rare population, existing only one in a million bone marrow cells [18]. Owing to stem cell’s self-renewal and reconstruction ability, in vivo transplantation into conditioned hosts has been applied for the characterization of these stem cells. HSCs have shown self-renewal and differentiation capacity even in a single-cell level [19, 20]. In mice, HSC activity is evaluated and quantified by transplanting cells into myeloablative (usually by irradiation), syngeneic recipients. Availability of severe combined immunodeficient (SCID) mice made it possible to apply this transplantation strategy to human materials such as bone marrow and cord blood. Development of severe immunodeficient nonobese diabetic/SCID (NOD/SCID) mice and its subsequent generation of NOD/SCID/IL-2Rg null (NOG or NSG) mice allowed engraftment of a very small number of cells and greatly enhanced the isolation of HSCs [21–24]. Cells that generate all lineages but are capable of engrafting only hematopoietic lineages transiently are defined as short-term HSCs (ST-HSCs) or multipotent progenitors (MPPs), whereas HSCs that repopulate hematopoietic lineages beyond 12 weeks are defined as long-term HSCs (LT-HSCs) [20].
Lineage and differentiation stage-specific expression of surface proteins is notable in hematopoietic cells; therefore, HSCs and progenitor cells are defined by the cell surface phenotype, as described in Fig. 1.2 [20]. Prospective isolation of highly purified HSCs is made possible using monoclonal antibodies for these cell surface proteins and by precise fluorescence-activated cell sorting (FACS) [25]. Mouse HSCs were initially isolated as lineage marker-negative (Lin−), c-kit+, and Sca-1+ (LSK) population [26, 27]. By extensively studied xenograft models, it was identified that a proportion in CD34− LSK cells, which show CD150+CD48−SLAM+ phenotype, possesses LT-HSCs activity in mouse [25, 28]. By contrast, CD34+ cells, which are found in less than 5% of hematopoietic cells, are enriched for HSCs and progenitors in humans. While phenotype of human HSCs was described as Lin−CD34+CD38−CD90(Thy1)+CD45RA− [21, 29–31], Lin−CD34+CD38−CD90−CD45RA−, which has lost CD90 expression, represents its descendant multipotent progenitors [32]. In addition, a recent study showed an essential role of CD49f in separation of HSCs [23]. Human HSCs are currently thought to be phenotypically Lin−CD34+CD38−CD90+CD45RA−CD49f+. When HSCs give rise to MPPs, first, CD49f expression is lost, and then they differentiate into immature progenitors and mature hematopoietic cells with specific cell surface phenotype under the influence of various transcriptional controls (Fig. 1.2) [20].
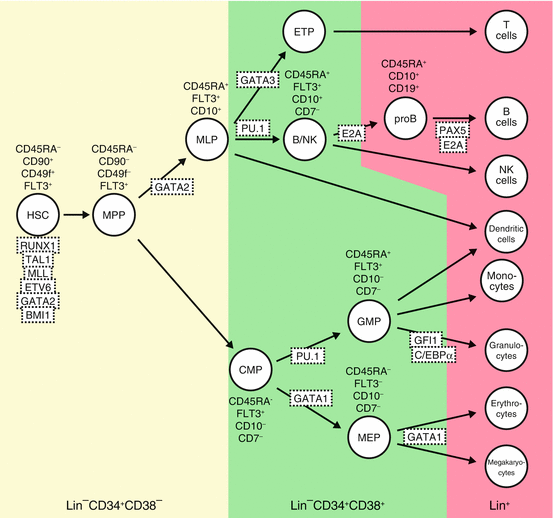
Fig. 1.2
Hematopoietic hierarchies and their regulators in human. Using specific antibodies and applying precise fluorescence-activated cell sorting (FACS) technique, HSCs, progenitor cells, and differentiated hematopoietic cells can be defined by cell surface phenotypes as shown. Transcription factors and signaling pathway known to be involved in hematopoietic cell differentiation are depicted. Hematopoietic development is blocked in the absence of many of these factors, as determined by gene knockout studies. MPPs multipotent progenitors, MLP multi-lymphoid progenitors, ETPs earliest thymic progenitors, CMPs common myeloid progenitors, GMPs granulocyte-macrophage progenitors, MEP megakaryocyte/erythroid progenitors, Lin cocktail of cell surface markers for all terminally differentiated hematopoietic cells. Modified from [20]
Within the CD34+CD38− population in human, CD90−CD45RA− cells are multipotent progenitors (MPP) with transient multilineage engraftment capacity [32]. CD90−CD45RA+ cells show differentiation to all lymphoid lineages as well as some of the myeloid lineage without self-renewal ability, functionally similar to murine lymphoid-primed multipotent progenitors (LMPP) [33] corresponding to multi-lymphoid progenitor (MLP) in human.
1.2.3 Transcriptional Control of HSCs: Hematopoietic Hierarchies and Lineage Determination
Blood cells are produced in developmental hierarchy, with multipotent HSCs at the apex and terminally differentiated cells at the bottom [20]. Key regulators controlling hematopoietic cell maintenance and differentiation differ in various stages of hematopoietic cell development, and many of these regulators are known to be transcription factors that play a central role in determining cell fate intracellularly. To date, more than 50 transcription factors have shown to affect HSCs survival and differentiation, forming transcriptional hierarchies active during hematopoietic differentiation (Fig. 1.2). Through the analysis of conventional gene knockout mice, some transcription factors have proven to play a crucial role in each stage of hematopoietic cell development. These crucial transcription factors have been often identified as targets of leukemia-specific gene abnormalities such as chromosome translocations, resulting in fusion gene formation.
HSCs and progenitor cells also respond to some external signals by activating a series of signaling cascades inside the cells, which may also activate other signaling pathways to form complex networks, finally activating lineage determinant transcription factor(s) [34, 35]. External signals, which work as determinants for stem cell fate decision, include interactions with surrounding cells through cell adhesion molecules and secreted molecules such as cytokines. Thus, microenvironments in the stem cell niche are critical for regulating the fate of HSCs [36, 37].
Growth factor receptor FLK1, as well as its ligand VEGF, and transcription factor TAL1 (SCL), with its protein partner LMO2, are essential factors for HSC formation and function in the early hematopoietic stage of development such as in the embryonic yolk sac. TAL1 and its protein partner LMO2 are necessary for the development of both primitive and definitive hematopoiesis because they specify blood lineage from the hemangioblast [38]. In AGM and fetal liver stage, AML1 (RUNX1), GATA2, GATA3, LMO2, TEL (ETV6) transcription factors, histone methyltransferase MLL (KMT2A), and KIT with its ligand SCF (KITLG) are required for hematopoietic cell survival, proliferation, and differentiation [35, 39–42]. ETS transcription factor TEL initiates adult hemangioblast program in the dorsal lateral plate mesoderm by switching on the FLK1 ligand VEGFA, where ETS factor FLI1 together with GATA2 ensures the expression of FLK1 [43].
The pluripotent HSCs generate mature hematopoietic cells via multipotent progenitors and committed precursors. Key regulators that control hematopoietic cell maintenance and differentiation differ in various stages of hematopoietic cell development (Fig. 1.2). During fate decision of hematopoietic cells, key lineage-restricted factors promote their own lineage differentiation and simultaneously act against factors that drive cells into other linages [35]. GATA1 promotes erythroid, megakaryocytic, and eosinophil differentiation, whereas PU.1 (SPI1) promotes myeloid differentiation in common myeloid progenitors (CMP). This GATA1 and PU.1 antagonism promotes cell differentiation to their specific lineage. This kind of lineage determination is also known between EKLF and FLI1 for erythroid and megakaryocytic lineages in megakaryocyte/erythroid progenitors (MEP), GFI1 and PU.1 for neutrophil versus monocyte differentiation in granulocyte/macrophage progenitors (GMP), and C/EBP and FOG1 for eosinophil and multipotent cell fate [35].
For B-cell development, PAX5 is required for B-cell commitment [44], and other B-cell transcription factors such as E2A and EBF1 specify gene activation toward the B-lineage. In T-cell development, NOTCH1 commits cells to T-cell lineage, whereas other transcription factors such as GATA3 support T-cell differentiation under NOTCH signaling. Further, GATA3 and T-bet (TBX21) antagonize in T-cell differentiation to promote Th1 and Th2 cells [45]. It is now known that these lineages can be reprogrammed upon expression of critical transcription factors such as GATA1, C/EBP, and GATA3 [35].
Notably, in HSCs and progenitor cells, alternative cell fate potentials are preserved by keeping simultaneous low expression of key genes for several lineages [46]. Differentiation of HSCs to certain lineage takes place by selecting and stabilizing the expression of a subset of genes, which are essential for that direction from a wide range of expression of lineage-oriented genes and, at the same time, downregulating expression of other irrelevant genes under the effects of selected dominantly expressed gene(s). Thus, to maintain a hierarchy of hematopoietic differentiation from HSCs, precise regulation of transcription factors expression is critical.
This multiple co-expression of lineage-oriented genes, such as transcription factors in HSCs and progenitor cells, is essential for maintaining multipotentiality and flexibility in cell fate decisions. Loss of expression of one lineage determinant transcription factor and upregulation of another lineage-specific gene in already lineage-committed cells can induce lineage conversion in normal hematopoiesis [46, 47] and also in leukemogenesis [48], indicating lineage plasticity.
1.2.4 Microenvironment of HSCs: The Niche
HSCs undergo a wide range of cell fate, such as self-renewal, quiescence, and differentiation into all mature blood cells. Fate of HSCs is tightly regulated by external stimuli provided by the HSC microenvironment, known as niche, and by intercellular regulatory programs. The adult bone marrow niche is best studied among various stages of fetal and adult hematopoietic development.
The major components of the niche that support HSC fate in the bone marrow are the osteoblastic and vascular niches, which lie in close proximity and are thought to influence HSCs in a cooperative manner (Fig. 1.3) [49].
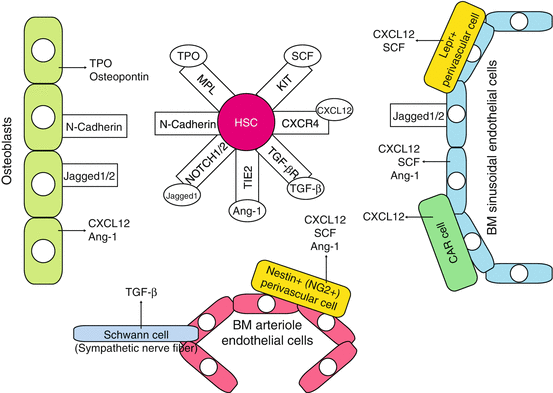
Fig. 1.3
Stem cell niche in adult bone marrow. The major components of the niche that support HSCs cell fate in the bone marrow are the osteoblastic and vascular niches that lie in close proximity and are thought to influence HSCs in a conjoined manner. HSCs exist adjacent to the osteoblasts, which align bone surface of the bone marrow, and are under the regulation of bone morphogenetic protein (BMP) (the osteoblastic niche). HSCs are also found to have migrated to blood vessels within the bone marrow—the vascular niche. Bone marrow also contains other stromal cells that support HSCs and hematopoietic progenitors by producing cytokines such as KIT ligand SCF. CAR cell CXCL12-abundant reticular cell, Ang-1 angiopoietin-1, OPN osteopontin
HSCs exist adjacent to the osteoblasts, which align bone surface of the bone marrow, and are under the regulation of bone morphogenetic protein (BMP)—the osteoblastic niche. Various studies using mouse models showed that osteoblastic niche maintains HSCs thorough various signaling molecules, including Notch1, Tie-2, N-cadherin, and Mpl [49]. HSCs are also found to migrate to blood vessels within the bone marrow—the vascular niche. LT-HSCs in mice are defined through SLAM markers (CD150+, CD41−, CD48−, cKit+, Sca-1+, lineage-) and lie adjacent to the bone marrow sinusoidal endothelial cells that express Notch ligands, Jagged-1 and Jagged-2, and KIT ligand SCF. The bone marrow also contains other stromal cells that support HSCs and hematopoietic progenitors by producing cytokines such as SCF. Cells surrounding the vasculature, CXC chemokine ligand 12 (CXCL12)-abundant reticular (CAR) cells, and Nestin+ mesenchymal stem cells have been identified as the independent niche components (the perivascular niche). CAR cells exert their niche functions through SCF and CXCL12 secretion into the microenvironment [49–51], regulating the proliferation of HSCs. Nestin+ cells possess the ability of multilineage differentiation into various mesenchymal cell lineages, including osteoblasts. Nestin+ cells highly express chemokine/cytokines such as CXCL12, SCF, angiopoietin-1, and osteopontin that are involved in the regulation of HSCs with other niche components. Bone marrow macrophages, non-myelinating Schwann cells [52], and extracellular matrix protein such as tenascin-C (TNC) [53] have been recently reported to possess niche function in the bone marrow.
1.3 Stem Cells in Abnormal Hematopoiesis
1.3.1 Disruption of Transcription Network Leads to Hematopoietic Malignancy
The transcription factors that are critical for hematopoiesis are mainly various classes of DNA-binding proteins, and perturbation of the function of these proteins is known to cause hematopoietic defect and malignancies. Many of these transcription factors involved in hematopoiesis are known to be disrupted by chromosome translocations, deletions, or mutations, resulting in defective normal cell differentiation and/or acquisition of cell proliferation, which consequently leads to leukemia or other hematopoietic malignancies. Leukemia-associated chromosome translocation often produces either chimeric fusion proteins with abnormal function or deregulated expression of certain genes that drives cell transformation. Some of the common examples of chimeric fusion proteins are MLL fusion proteins such as MLL-AF9, TEL (ETV6)-associated fusion proteins such as TEL-AML1 (ETV6-RUNX1), and AML1 (RUNX1)-associated fusion proteins such as AML1-ETO (RUNX1-RUNX1T1), which are thought to affect normal HSC development leading to leukemia. Examples of chromosome translocation contributing to leukemogenesis by causing aberrant expression of transcription factors are TAL1 (SCL) and LMO2 in T-cell acute leukemia, caused by translocation with TCR genes. These aberrant transcription factors generated by the chromosome translocation cause improper activation or repression of their target genes and inhibit function of other critical factors, exerting multiple downstream effects that lead to aberrant hematopoiesis. Somatic mutation in transcription factors itself such as PU.1 and C/EBPα (CEBPA) is also known to cause myeloid leukemia [54–56]; mutations in PAX5 [57–59] and other B-cell factors lead to B-cell leukemia, and NOTCH1 mutation leads to T-cell leukemia [60].
1.3.2 Cancer Stem Cells and Xenograft Transplantation Models
It is widely accepted nowadays that most tumors, including leukemia, are sustained by a subpopulation of the self-renewing stem cells, called cancer stem cells (CSCs). The presence of these CSCs maintain the clonal architecture of cancer itself, and in clinical terms, failure to eradicate CSCs by therapy definitely causes post-therapy relapse, i.e., reconstruction of clonal architecture of cancer by CSCs.
By definition, CSC is a cell within a tumor that possesses the ability to self-renew and simultaneously produce heterogeneous lineages of cancer cells that comprise the tumor [61]. Therefore, CSCs can only be defined experimentally by demonstrating their ability to recapitulate the generation of a continuously growing tumor. This ability is best assayed using xenotransplantation models and measuring tumor formation in serially transplanted recipients [2, 19, 61, 62]. To prove the self-renewal ability of CSCs, isolated CSCs from tumor formed in the first recipient animal should generate tumor in the second recipient and continuously generate original tumor in the succeeding recipients (serial transplantation). Tumors generated in the recipient animal should maintain the heterogeneity observed in the original tumor, containing descendant cells without self-renewal ability. In this experimental system, CSCs are termed as “tumor-initiating cell (T-IC)” or leukemia-initiating cell (L-IC) in case of leukemia. AML represents the very first example of human cancers from which CSCs have been identified as SCID leukemia-initiating cell (SL-IC) [1].
Therefore, description of subpopulation of cells that sustain whole cancer cells as CSCs or LSCs is basically conceptual, in contrast to xenotransplant assay-based term T-IC or L-IC. Although these terms are considered to be largely overlapping, consistency between these two terms from different standpoints is currently a key issue [2, 62].
1.3.3 Preleukemic Stem Cells as the Reservoir of LSCs
Development of leukemia, like that of other cancers, is a multi-step process based on the consecutive acquisition of several genetic changes such as driver mutations. Generally, a single driver mutation is not enough to cause leukemia, and at least another cooperating mutation is necessary for the initiation to the formation of LSCs. There are animal models showing that some leukemia-specific fusion such as TEL-AML1 or AML-ETO is not sufficient to drive leukemia by themselves, and additional genetic hit(s) are necessary for generation of leukemia [63–67]. Therefore, on the pathway of leukemogenesis, preleukemic stem cells (pre-LSCs) and their descendant preleukemic cells, which do not harbor enough number of driver mutations, are present before proceeding to LSCs. In accord with this, expression of some leukemia-specific fusion genes is detectable in nonleukemic blood samples. The TEL-AML1 and AML1-ETO fusion genes present in the cord blood are reported to be 10−3 to 10−4 in frequency, suggesting the presence of cells with significant ability of clonal expansion and indicative of preleukemic nature [68–70]. In addition, there are reports showing that not only TEL-AML1 and AML1-ETO but also BCR-ABL1 and BCL2-IGH fusion genes exist in a detectable amount in the blood cells of normal adults [71–75], indicating that the formation of preleukemic cells by fusion genes is a fairly common phenomenon. By contrast, frequency of TEL-AML1 and the amount of TEL-AML1-positive cells in a healthy individual are reported to decrease with age, indicating that preleukemic TEL-AML1-positive clones do not persist in adulthood [76].
As for MLL-AF4, functional significance in normal individual is doubtful. Detection of MLL-AF4 fusion gene by RT-PCR at high frequency (~25%) in the fetal liver and fetal bone marrow has been reported [77], but these findings were not subsequently confirmed by others. Although MLL-AF4-positive leukemia is found in high frequency in infant leukemia and is proven to be formed in utero, it is not detected in normal individual, which suggests that single MLL-AF4 fusion may give rise to leukemic stem cell by itself, and preleukemic stage does not exist or exists only for a very short period. By contrast, there are controversial results from murine models where MLL fusion gene requires protracted latency before leukemic transformation [78, 79]. Additional studies are needed to draw any conclusion regarding the functional role of MLL-AF4 in preleukemic and leukemic stem cells.
In AML1-ETO-positive AML, it is known that the fusion gene can be detected during remission and can also exist in the normal hematopoietic stem cell compartment [80–82]. As this AML1-ETO-positive hematopoietic stem cell possesses the ability to differentiate not only to myeloid lineage but also to B-cell lineage, the existence of preleukemic stem cell is suggested. NPM1 and TET2 mutations, detected in high frequency in AML, are reported to exist during remission and also in the normal hematopoietic stem cell compartment, similar to that in AML1-ETO [83–85]. These cells do not possess second genetic changes such as FTL3 internal tandem duplication (FLT3-ITD), indicating preleukemic potential of these cells.
The same phenomenon was observed in AML with DNMT3A and IDH1/IDH2 mutations [85]. These leukemogenic driver mutations were persistently present in the patients’ blood in remission. Since co-occurring mutations such as those in NPM1 or FLT3 were not present, these cells should be considered preleukemic, without the ability to fully transform to cause overt leukemia. In addition, DNMT3A mutation was also detectable in non-myeloid lineage cells, indicating multipotent HSC origin. HSCs with DNMT3A mutations persistently and clonally expand more effectively than normal HSCs, indicating retention of advanced self-renewal ability. These cells can be called preleukemic stem cells (pre-LSCs).
Persistence and limited expansion of TEL-AML1-positive cells for several years before reaching the diagnosis of ALL, and persistence of cells with AML1-ETO, DNMT3A, or IDH1/IDH2 mutations in remission supported the presence of functional pre-LSCs. These pre-LSCs may be resistant to chemotherapy and work as a reservoir to generate another leukemic clone at relapse [86].
1.3.4 In Utero Generation of Pre-LSCs/LSCs in Childhood Leukemia
It is well recognized nowadays that leukemic cells in some of the childhood leukemia arise in utero [87, 88], and it was first identified by the studies of monozygotic twins with concordant leukemia, carrying identical genetic marker for chimeric fusion genes formed in utero [87, 89]. This was followed by retrospective studies using neonatal blood spots, demonstrating that common fusion genes such as MLL-AF4 in infant ALL, TEL-AML1 in childhood ALL, and AML1-ETO in childhood AML arise predominantly in utero and are present before and at the time of birth in circulating blood. In utero, origin of leukemic stem cell is proven not only in infant or childhood B-ALL with TEL or MLL fusion genes but also in ALL with high hyperdiploidy [90], T-ALL with NOTCH1 mutation [91], and AML with AML1-ETO [92] usually occurring later in the childhood.
This indicates that the fusion gene-positive cells represent expanded preleukemic clones, which will remain pathologically and clinically silent in the absence of additional postnatal second genetic hits, according to the classical “two-hit” model of Knudson for noninheritable cancer in childhood (Fig. 1.4) [69, 93]. The presence of these nonleukemic, preleukemic clones is also supported by the observation of TEL-AML1 and AML1-ETO fusion genes, which were shown to exist in the cord blood at a frequency of 1%, almost 100-fold higher than the actual risk of clinically diagnosed leukemia with the same fusion gene.
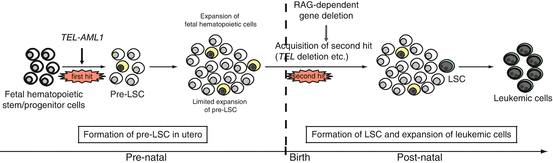
Fig. 1.4
In utero origin of ALL and multi-step leukemogenesis. A hypothesis for preleukemic and leukemic stem cell formation in TEL-AML1-positive ALL. A preleukemic stem cell (pre-LSC) is generated by the acquisition of TEL-AML1 translocation, but the cell is not completely transformed. Overt leukemia develops when one or more additional genetic changes (i.e., second hit, third hit, and so on) occur in the pre-LSCs after a relatively long latency period. The commonly observed secondary genetic changes are thought to be induced by recombination activating gene (RAG)-dependent gene deletions, which may explain why LSCs exist only in B-cell precursors and not in immature hematopoietic cells in TEL-AML1-positive ALL. Note that the same hypothesis may be applied to ALL with MLL-AF4, high hyperdiploidy, NOTCH1 mutation, and in some type of AML such as AML1-ETO (which also originate in utero). The cells in yellow indicate pre-LSCs that are formed as a result of the first genetic change. The cells in green indicate LSCs that acquire proliferative capacity by additional genetic change(s)
1.3.5 Genetic Profiles of Leukemic Stem Cells and Their Progeny
In the past decade, advances in next-generation sequencing technique have resulted in an enormous amount of newly identified genetic abnormalities to better understand the pathogenesis and etiology of malignant transformation of HSCs. In colon and breast cancer, 50–80 genetic changes were detected in the coding region of associated genes [94], whereas in AML and ALL, these genetic changes were much less compared to that in most other adult cancers: only around ten mutations in average [95–99]. Excluding common genetic changes such as FLT3, NPM1, and NRAS mutation (called driver mutation), the rest is not recurrent and thought to contribute in determining characteristic of the cells rather than in cell transformation itself (i.e., “passenger” mutation). Preleukemic cells, acquiring various random genetic and epigenetic changes, transform into leukemic stem cells, following the initiating events that are crucial for cell transformation.
There are several questions raised with respect to cell transformation by genetic changes. First, what is the cell of origin where leukemia arises? Next, how many and what kind of genetic changes are required for leukemic transformation? Finally, what clinical implications can we learn from the biology of leukemic stem cells? The origin and evolution of genetic mutation have been studied thoroughly in acute promyelocytic leukemia (APL, FAB M3 subtype) or AML without maturation (FAB M1 subtype) with normal karyotype by performing whole genome sequencing [98]. They found that the number of recurrent mutations required to generate malignant founding clone is three in M1 subtype and two in M3 subtype (one of which is PML-RARA). Other additional mutations obtained in these founder clones are cooperating mutations, which contribute to disease progression and/or relapse. This result is in accord with the previous suggestion that introduction of class I mutation that cause cell proliferation and class II mutation that results in block in differentiation is sufficient for cell transformation [100, 101].
In ALL, SNP array and whole exome or whole genome analysis revealed that TEL-AML1-positive leukemic cells do not often carry mutations in tyrosine kinase. By contrast, deletions in genes associated with B-cell development, such as TEL, CDKN2A, BTG1, PAX5, and RAG2, were frequent, suggesting that gene deletion mainly contributes to cell transformation in TEL-AML1-positive ALL. The number of additional genetic changes required in cell transformation is six in average in TEL-AML1-positive ALL and one in MLL fusion-positive ALL. These results suggest that although some variation exists among fusion genes, the average number of mutations required for transforming preleukemic cell to leukemic stem cells is one or two in average.
1.3.6 Clonal Evolution of Preleukemic Cells to Leukemic Stem Cells
Pre-LSC is generated from hematopoietic stem/progenitor cell by acquisition of one driver mutation which could provide or enhance its self-renewal/survival capacity. Transition from pre-LSC to LSC, which gives rise to overt leukemia, usually needs acquisition of another driver mutation that can complete the leukemogenic transformation. Acquisition of mutation(s) usually occurs randomly in any gene(s) and is essential to have effective mutation in proper gene that can act as a leukemogenic driver for the generation of LSC. Because of this random, continuous acquisition of gene mutation(s), there is always a possibility that descendant of single pre-LSC clone gets different driver mutation producing different subclones of LSC in a single patient. Same phenomenon can also happen in each LSC clone resulting in further generation of different subclones of LSCs with different characteristics such as chemoresistance, phenotype, and so on. This clonal evolution, due to genomic diversity, is universally observed in cancer including leukemia [102] and among several LSC clones. LSC with outstanding growth is selected and becomes a dominant clone within leukemic cell population. The presence of these genetically distinct LSC clones often causes a clonal change at relapse or during chemotherapy.
1.4 Characteristics of Leukemic Stem Cells
1.4.1 Leukemic Stem Cells in AML
AML is an aggressive disease predominantly seen in adult, which represents poor prognosis of 30–40% 5-year overall survival. In order to study the cell of origin of leukemia, surface antigen has been used as markers to separate and enrich the LSC subset from the leukemic population by using technological advanced fluorescence-activated cell sorting (FACS). Regardless of the heterogenetic characteristics of the leukemic blasts, CD34+CD38− phenotype similar to normal HSCs was originally defined as LSC [1] in all subtypes of AML (except APL). This phenotype was identified as cells with self-renewal and proliferative capacities that cause leukemia (L-ICs) in NOD/SCID mice, technically termed as SCID leukemia-initiating cell (SL-IC). Like the normal hematopoietic system, leukemic clone is organized as a hierarchy in AML with LSCs among the CD34+CD38− cells on the apex (Fig. 1.5). The number of LSCs in AML varies among patients but is estimated to be approximately 0.2–200 in a million AML cells [1].
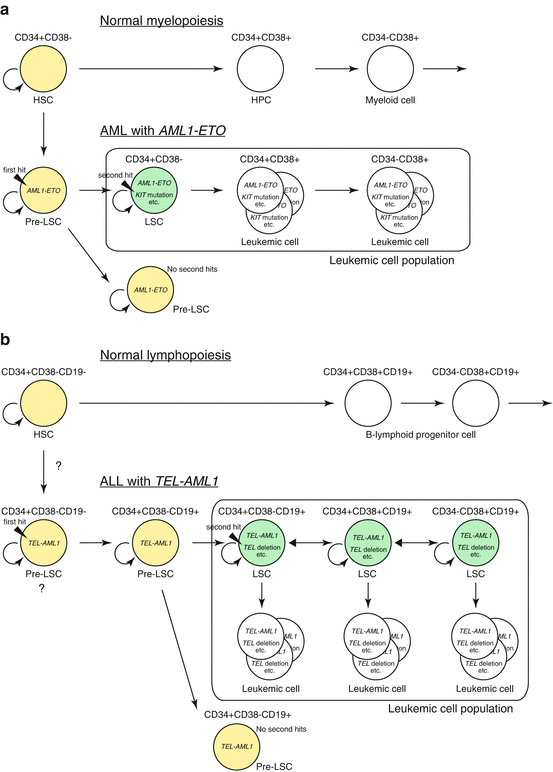
Fig. 1.5
Leukemic stem cell in AML and ALL. (a) Hierarchy of leukemic stem cells (LSCs) in AML. AML with AML1-ETO fusion is shown as an example. Pre-LSCs are formed as a result of the first genetic change (i.e., generation of AML1-ETO). There is no overt leukemia unless pre-LSCs acquire secondary genetic changes to form LSCs. LSCs exist in immature CD34+CD38− population that can give rise to its progeny and accounts for majority of leukemic cells. The cells in yellow indicate dormant cells that have self-renewal capacity but lack robust proliferation capacity, such as normal hematopoietic stem cells (HSCs) and pre-LSCs. The cells in green indicate LSCs that have acquired proliferative capacity and can thus induce leukemia. The cells in white within the box are the majority of leukemic cells that does not possess self-renewing capacity to induce leukemia on its own. HPC hematopoietic progenitor cell. (b) Stochastic formation of ALL stem cells. ALL with TEL-AML1 fusion is shown as an example. In childhood ALL, pre-LSCs (shown in yellow) are formed by the acquisition of the first genetic change, i.e., the formation of TEL-AML1 fusion gene, which may occur at the hematopoietic stem cell level. Pre-LSCs possibly differentiate into aberrant CD34+CD38−CD19+ cells that are not present during normal hematopoiesis. Pre-LSCs then acquire a second genetic change during various stages of differentiation to form LSCs (shown in green) that have the ability to dedifferentiate and self-renew. LSCs are reported to account for 1–24% of total leukemic cells in ALL, and each LSC not only self-renew but also produce progeny that does not possess self-renewing ability (shown in white), resulting in the development of clinically overt leukemia. These LSCs may differentiate or dedifferentiate, making the mixed population of various phenotypes. The accurate stage of cell development where first and second hits are acquired is currently unknown
LSCs of CD34+CD38− phenotype in AML can be further divided into CD45RA+CD90− and CD45RA−CD90− populations. These cells can be derived from normal HSCs population that possesses Lin−CD34+CD38−CD45RA−CD90+ phenotype. CD45RA+CD90− in LSCs resembles LMPP (lymphoid-primed multipotent progenitor), whereas CD45RA−CD90− resembles GMP (granulocyte-macrophage progenitor) in normal hematopoietic development. In leukemic hierarchy, LMPP-like cells (equivalent of MLP in normal hematopoietic development) stay primitive to GMP-like cells within LSC fraction [33].
While L-ICs in SCID xenotransplant assay were enriched in the Lin−CD34+CD38− fraction compared with CD38+ and the other fractions analyzed as originally reported, L-ICs in Lin−CD34+CD38− fraction represented only one-third, at the maximum, of all L-ICs present in the whole specimen in most of the AML cases. These results indicate that human LSCs in AML are enriched in Lin−CD34+CD38− fraction, sharing the phenotype with normal primitive hematopoietic cells but are not restricted to this fraction with immature phenotype suggesting a plasticity of the cancer stem cell phenotype [103].
In AML, while LSCs may arise from normal HSCs by acquiring leukemogenic driver mutation(s), some of the LSCs may arise from the differentiated, myeloid-committed progenitors by recovering self-renewal capacity, as a result of obtained driver mutation(s). Some driver mutations do not have transforming potential unless they occur in hematopoietic stem cells. Expression of BCR-ABL1 fusion gene must occur in HSCs to cause CML in murine models, providing evidence that LSCs reside at the HSC level [104]. As HSCs originally have self-renewing capacity, AML1-ETO, which blocks cell differentiation, can transform HSCs into LSCs with other driver mutations such as RAS and FLT3 and provide growth advantage. On the other hand, MLL-ENL [105], MOZ-TIF2 [104], MLL-AF9 [106], and PML-RARA [107, 108] can provide self-renewing capacity to the cells, initiating leukemia even when it occurred in more lineage-restricted progenitors.
In case of AML1-ETO-positive AML, isolated CD34+CD90−CD38− cells with AML1-ETO fusion give rise to multilineage progenitor with normal differentiation capacity as shown in vitro hematopoietic progenitor assay, whereas more mature CD34+CD90−CD38+ with AML1-ETO form blast colonies [82]. These results indicate that AML1-ETO is formed in primitive stem cell, but subsequent events occur in committed progenitor level, giving rise to LSCs.
Not much is known about LSCs of childhood AML. From LSCs with disease-type-specific fusion genes, such as AML1-ETO, one can speculate that the rules obtained from adult AML can be applied also in childhood AML. On the other hand, mutation in epigenetic regulator, such as DNMT3A, IDH1, and IDH2, are rare in childhood AML [109]. It has been reported that gene mutation in HSCs accumulate with age and that most of the mutations present in AML genomes were already present in the hematopoietic cell that was “transformed” by the initiating mutation in adult AML [98]. As young HSCs do not carry as much number of mutations as the old HSCs, some mechanism may vary in transformation of the young HSCs.
1.4.2 Leukemic Stem Cells in Acute Lymphoid Leukemia (ALL)
ALL is the most common hematologic malignancy in childhood. Generally leukemic cells respond well to combination chemotherapy resulting in over 80% of 5-year event-free survival in children.
Leukemic stem cells in ALL was first described in BCR-ABL1-positive ALL, and similar to AML, L-IC activity was identified in immature Lin−CD34+CD38− population by NOD/SCID xenotransplantation assay [110]. However, several studies showed different views about L-IC activity in ALL.
TEL-AML1 is the most frequent fusion gene in childhood ALL formed as a result of chromosome translocation t(12;21)(p13;q22). So far, several xenotransplantation studies of TEL-AML1-positive ALL were reported with varying results. Early report indicated that, in at least some of the ALL patients with TEL-AML1, CD34+CD19− cells, but not CD19+ cells, showed L-IC activity [111]. However, later report contradicted those early results and showed that CD19+ cells are the main source of L-IC activity [112–114]. Some reports even denied the presence of L-IC activity in CD34+CD19− cells [113]. FISH (fluorescence in situ hybridization) analysis of sorted leukemic cells partly supported the absence of L-IC activity in immature CD34+CD19− cells in TEL-AML1-positive ALL and also showed an absence of TEL-AML1 fusion on CD34+CD19− cell population [115].
As aforementioned, TEL-AML1 is frequently (almost always) generated in utero, and TEL-AML1-harboring hematopoietic cells exist as preleukemic (stem) cells for several years until overt leukemia develops after acquisition of additional genetic mutations sufficient for transformation. In the detailed study of twin leukemia case, TEL-AML1-positive preleukemic cells existed in CD34+CD38−CD19+CD10− population and were also present in nonleukemic twin as well as the leukemic twin [114]. These findings are also comparable with LSCs in CD19+ cell population in TEL-AML1-positive ALL, suggesting CD19+ B-lineage-committed cells as the target for the leukemogenic transformation by TEL-AML1 fusion.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


