Abstract
A variety of systemic illnesses including acute and chronic infections, neoplastic diseases, connective tissue disorders, and storage diseases are associated with hematologic manifestations.
Keywords
Systemic illness, hematology, iron deficiency, anemia, bone marrow, organ
A variety of systemic illnesses including acute and chronic infections, neoplastic diseases, connective tissue disorders, and storage diseases are associated with hematologic manifestations. The hematologic manifestations are the result of the following mechanisms:
- •
Bone marrow dysfunction
- •
Anemia or polycythemia
- •
Thrombocytopenia or thrombocytosis
- •
Leukopenia or leukocytosis
- •
- •
Hemolysis
- •
Immune cytopenias
- •
Alterations in hemostasis
- •
Acquired inhibitors to coagulation factors
- •
Acquired von Willebrand disease
- •
Acquired platelet dysfunction
- •
- •
Alterations in leukocyte function
Hematologic Manifestations of Diseases Related to Various Organs
Heart
Microangiopathic hemolysis occurs with prosthetic valves or synthetic patches utilized for correction of cardiac defects, particularly when there is failure of endothelialization (“Waring Blender” syndrome) or rarely after endoluminal closure of a patent ductus arteriosus. Microangiopathic hemolysis has the following characteristics:
- •
Hemolysis is secondary to fragmentation of the red cells as they are damaged against a distorted vascular surface.
- •
Hemolysis is intravascular and may be associated with hemoglobinemia and hemoglobinuria.
- •
Iron deficiency occurs secondary to the shedding of hemosiderin within renal tubular cells into the urine (hemosiderinuria).
- •
Thrombocytopenia secondary to platelet adhesion to abnormal surfaces.
- •
Autoimmune hemolytic anemia may occasionally occur after cardiac surgery with the placement of foreign material within the vascular system.
Cardiac Anomalies and Hyposplenism
Cardiac anomalies, particularly situs inversus , may be associated with hyposplenism and the blood smear may show Howell–Jolly bodies, Pappenheimer bodies, and elevated platelet counts.
Infective Endocarditis
Hematologic manifestations include anemia (due to immune hemolysis or chronic infection), leucopenia, or leukocytosis and rarely thrombocytopenia and pancytopenia.
Coagulation Abnormalities
- •
A coagulopathy exists in some patients with cyanotic heart disease. The coagulation abnormalities correlate with the extent of the polycythemia. Hyperviscosity may lead to tissue hypoxemia, which could trigger disseminated intravascular coagulation (DIC).
- •
Marked derangements in coagulation such as DIC, thrombocytopenia, thrombosis, and fibrinolysis can accompany surgery involving cardiopulmonary bypass. Heparinization must be strictly monitored.
Platelet Abnormalities
Quantitative and qualitative platelet abnormalities are associated with cardiac disease:
- •
Thrombocytopenia occurs secondary to microangiopathic hemolysis associated with prosthetic valves.
- •
Cyanotic heart disease can produce polycythemia, thrombocytopenia, prolonged bleeding time, and abnormal platelet aggregation.
- •
Patients with chromosome 22q11.2 deletion (DiGeorge syndrome) can have platelet abnormalities including the Bernard–Soulier-like syndrome due to haploinsufficiency of the gene for GP1BB and thrombocytopenia due to autoimmunity.
Polycythemia
- •
The hypoxemia of cyanotic heart disease produces a compensatory elevation in erythropoietin and secondary polycythemia.
- •
Patients are at increased risk for cerebrovascular accidents secondary to hyperviscosity.
- •
Patients are also at risk for symptomatic hypoglycemia (especially in the neonatal period).
- •
The use of partial exchange transfusion has been suggested, although the long-term value of exchange has been challenged.
Gastrointestinal Tract
Esophagus
- •
Iron-deficiency anemia may occur as a manifestation of gastroesophageal reflux.
- •
Endoscopy may be required in unexplained iron deficiency.
Stomach
- •
The gastric mucosa is important in both vitamin B 12 and iron absorption.
- •
Chronic atrophic gastritis produces iron deficiency. There may be an associated vitamin B 12 malabsorption.
- •
Gastric resection may result in iron deficiency or in vitamin B 12 deficiency due to lack of intrinsic factor.
- •
Zollinger–Ellison syndrome (increased parietal cell production of hydrochloric acid) may cause iron deficiency through mucosal ulceration.
- •
Helicobacter pylori infection, in addition to causing chronic gastritis, has been implicated in the initiation of iron-deficiency anemia, vitamin B 12 deficiency, autoimmune thrombocytopenia, and platelet aggregation defects (ADP-like defect).
Small Bowel
- •
Celiac disease or tropical sprue may cause malabsorption of iron and folate. Table 2.1 lists the various hematologic manifestations of celiac disease.
Table 2.1
Hematologic Manifestations of Celiac Disease
Problem
Frequency
Comments
Anemia: iron deficiency, folate deficiency, vitamin B 12 deficiency, and other nutritional deficiencies
Common
The anemia is most commonly secondary to iron deficiency but may be multifactorial in etiology. Low serum levels of folate and vitamin B 12 without anemia are frequently seen. Anemia due to other deficiencies appears to be rare
Thrombocytopenia
Rare
May be associated with other autoimmune phenomena
Thrombocytosis
Common
May be secondary to iron deficiency or hyposplenism
Thromboembolism
Uncommon
Etiology is unknown but may be related to elevated levels of homocysteine or other procoagulants
Leukopenia/neutropenia
Uncommon
Can be autoimmune or secondary to deficiencies of folate, vitamin B 12 , or copper
Coagulopathy
Uncommon
Malabsorption of vitamin K
Hyposplenism
Common
Rarely associated with infections
IgA deficiency
Common
May be related to anaphylactic transfusion reactions
Lymphoma
Uncommon
The risk is highest for intestinal T-cell lymphomas
From: Halfdanarson et al. (2007) , with permission.
- •
Inflammatory bowel disease (IBD) may cause anemia of chronic inflammation and iron deficiency from blood loss.
- •
Eosinophilic gastroenteritis can produce peripheral eosinophilia.
- •
Diarrheal illnesses of infancy can produce life-threatening methemoglobinemia.
Lower Gastrointestinal Tract
- •
Ulcerative colitis is often associated with iron-deficiency anemia.
- •
Peutz–Jeghers syndrome (intestinal polyposis and mucocutaneous pigmentation) predisposes to adenocarcinoma of the colon.
- •
Hereditary hemorrhagic telangiectasia (Osler–Weber–Rendu disease) may produce iron deficiency, platelet dysfunction, and hemostatic defects.
Pancreas
- •
Hemorrhagic pancreatitis produces acute normocytic, normochromic anemia. It may also be associated with DIC.
- •
Shwachman–Diamond syndrome (SDS; see Chapter 13 ) is characterized by congenital exocrine pancreatic insufficiency, metaphyseal bone abnormalities, and neutropenia. There may also be some degree of anemia and thrombocytopenia.
- •
Cystic fibrosis produces malabsorption of fat-soluble vitamins (e.g., vitamin K) with impaired prothrombin production.
- •
Pearson syndrome is characterized by exocrine pancreatic insufficiency and severe sideroblastic anemia (see Chapter 13 ).
Liver
Anemia
Anemias of diverse etiologies occur in acute and chronic liver disease. Red cells are frequently macrocytic (mean corpuscular volume (MCV) of 100–110 fl). Target cells and acanthocytes (spur cells) are frequently seen. Some of the pathogenic mechanisms of anemia include:
- •
Shortened red cell survival and red cell fragmentation (spur cell anemia) in cirrhosis often occur in later-stage cirrhosis in the presence of dyslipidemia.
- •
Hypersplenism with splenic sequestration in the presence of secondary portal hypertension.
- •
Iron-deficiency anemia secondary to blood loss from esophageal varices in portal hypertension.
- •
Chronic hemolytic anemia in Wilson disease secondary to copper accumulation in red cells. Hemolytic anemia may be the presenting symptom in this disease.
- •
Aplastic anemia following acute hepatitis (usually seronegative) in certain immunologically predisposed hosts.
- •
Megaloblastic anemia secondary to folate deficiency in malnourished individuals.
Coagulation Abnormalities
The liver is involved in the synthesis of most of the coagulation factors. Liver dysfunction can be associated with either hyper- or hypocoagulable states because both procoagulant and natural anticoagulant synthesis are impaired. Table 2.2 lists the various coagulation abnormalities seen in liver disease and Table 2.3 lists the tests to differentiate between the coagulopathy of liver disease and other etiologies.
| Hemorrhage | Thrombosis |
|---|---|
| (1) Thrombocytopenia/platelet dysfunction due to hypersplenism, altered TPO production | (1) Decreased anticoagulant—AT-III Protein C and S |
| (2) Decreased liver synthesis of procoagulant factors | (2) Portal hypertension-portal vein thrombosis |
| (3) Impaired carboxylation of vitamin K factors | |
| (4) Dysfibrinogenemia | |
| (5) Hyperfibrinolysis due to increased tPA and decreased PAI, α 2 antiplasmin |
| Procoagulant factors | Liver | Vitamin K | DIC |
|---|---|---|---|
| F V | Decreased (late) | Normal | Decreased |
| F VII | Decreased (early) | Decreased | Decreased |
| F VIII | Normal/increased | Normal | Decreased |
Factor I (Fibrinogen)
Fibrinogen levels are generally normal in liver disease. Low levels may be seen in fulminant acute liver failure.
Factors II, VII, IX, and X (Vitamin K-Dependent Factors)
These factors are reduced in liver disease secondary to impaired synthesis. Factor VII is the most sensitive.
Factor V
Factor V does not require vitamin K for synthesis and is highly representative of actual liver function. Factor V levels at 36 h post liver injury have been used as a standalone marker for the possible need for transplant in patients with early liver failure.
Factor VIII
The procoagulant activity of Factor VIII is generally normal in liver disease. This makes Factor VIII an important factor to measure in distinguishing between DIC and severe liver disease in a patient with abnormal coagulation tests and thrombocytopenia. If there is associated DIC, factor VIII will be markedly depressed, whereas in severe liver disease Factor VIII remains close to or normal. Traditionally, the Factor VII and Factor VIII levels are measured along with the PT, PTT, and fibrinogen to distinguish liver disease from DIC.
Protein C, Protein S, and Antithrombin III
These natural anticoagulants are decreased in liver disease. Proteins C and S are most sensitive to vitamin K deficiency. In many cases this fall in the levels of natural anticoagulant creates a sensitive balance between loss of procoagulant activity and natural anticoagulant activity. Bleeding or thrombosis may appear quickly when additional illness (e.g., infection) may upset the balance.
Tissue plasminogen activator (TPA) and alpha-2-antiplasmin
Tissue plasminogen activator is cleared by the liver and as liver disease progresses TPA activity increases. Alpha-2-antiplasmin is also suppressed by liver disease, creating increased plasmin activity and ultimately the syndrome of hyperfibrinolysis with a tendency toward severe bleeding.
α 2 -Macroglobulin and plasmin activator inhibitor
These opponents of plasma activity are still present in liver disease.
Kidneys
Renal disease may affect red cells, white cells, platelets, and coagulation.
Severe renal disease with renal insufficiency is frequently associated with chronic anemia (and sometimes pancytopenia). This type of anemia is characterized by:
- •
Hemoglobin as low as 4–5 g/dl
- •
Normochromic and normocytic red cell morphology unless there is associated microangiopathic hemolytic anemia (as in the hemolytic-uremic syndrome (HUS)), in which case schistocytes and thrombocytopenia are seen
- •
Reticulocyte count low
- •
Decreased erythroid precursors in bone marrow aspirate.
The following mechanisms are involved in the pathogenesis of this type of anemia:
- •
Erythropoietin deficiency is the most important factor (90% of erythropoietin synthesis occurs in the kidney)
- •
Shortened red cell survival is secondary to uremic toxins or in HUS secondary to microangiopathic hemolysis
- •
Renal failure itself inhibits erythropoiesis and in conjunction with decreased erythropoietin levels produces a hypoplastic marrow
- •
Increased blood loss from a hemorrhagic uremic state and into a hemodialysis circuit causes iron deficiency.
- •
Dialysis can lead to folic acid deficiency.
Treatment
- •
Recombinant human erythropoietin (rHuEPO): 1
1 Thrombosis of vascular access occurs in 10% of cases treated with rHuEPO.
- •
Determine the baseline serum erythropoietin and ferritin levels prior to starting rHuEPO therapy. If ferritin is less than 100 ng/ml, give ferrous sulfate 6 mg/kg/day aimed at maintaining a serum ferritin level above 100 ng/ml and a threshold transferrin saturation of 20%. With the advent of less immunoreactive forms of intravenous iron, prophylactic strategies utilizing intravenous iron infusion at the end of dialysis have been very successful. These are usually well tolerated compared to oral iron on a daily basis.
- •
Start with rHuEPO treatment in a dose of 50–100 units/kg/day subcutaneously (SC) three times a week.
- •
Monitor blood pressure closely (increased viscosity produces hypertension in 30% of cases) and perform complete blood count (CBC) weekly.
- •
Titrate the dose:
- –
If no response, increase rHuEPO up to 300 units/kg/day SC three times a week
- –
If hematocrit (Hct) reaches 40%, stop rHuEPO until Hct is 36% and then restart at 75% dose
- –
If Hct increases very rapidly ( > 4% in 2 weeks), reduce dose by 25%.
- –
Figure 2.1 shows a flow diagram, in greater detail, for the use of erythropoietin-stimulating agents in patients with chronic kidney disease.
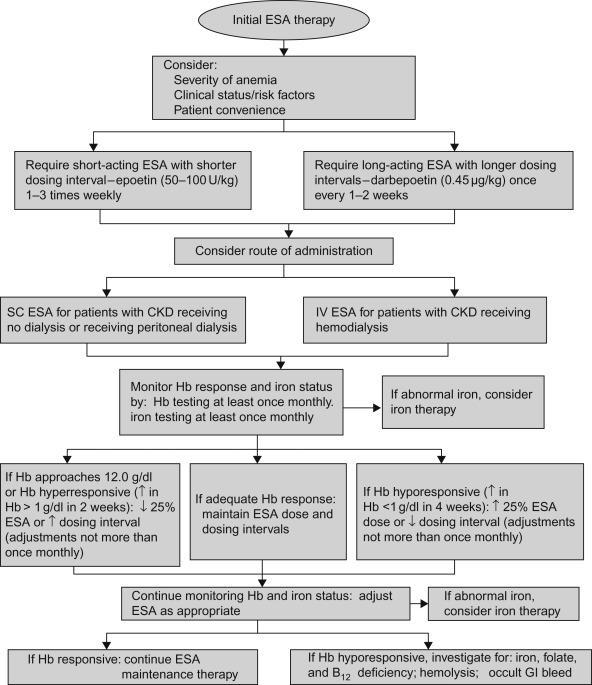
Figure 2.1
Recommended erythropoietin-stimulating agent (ESA) treatment in patients with chronic kidney disease. SC, subcutaneous; IV, intravenous; CKD, chronic kidney disease; ↑, increase; ↓, decrease.
Source: From Wish and Coyne (2007) , with permission.
- •
- •
Folic acid 1 mg/day is recommended because folate is dialyzable
- •
Packed red cell transfusion is rarely required.
Endocrine Glands
Thyroid
Anemia is frequently present in hypothyroidism. It is usually normochromic and normocytic. The anemia is sometimes hypochromic because of associated iron deficiency and occasionally macrocytic because of vitamin B 12 deficiency. The bone marrow is usually fatty and hypocellular and erythropoiesis is usually normocytic. The finding of a macrocytic anemia and megaloblastic marrow in children with hypothyroidism should raise the possibility of an autoimmune disease with antibodies against parietal cells as well as against the thyroid, leading to vitamin B 12 deficiency (juvenile pernicious anemia with polyendocrinopathies).
Adrenal Glands
- •
Androgens stimulate erythropoiesis.
- •
Conditions of androgen excess, such as Cushing syndrome and congenital adrenal hyperplasia, can produce secondary polycythemia.
- •
In Addison disease, some degree of anemia is also present but may be masked by coexisting hemoconcentration. The association between Addison disease and megaloblastic anemia raises the possibility of an inherited autoimmune disease directed against multiple tissues, including parietal cells (juvenile pernicious anemia with polyendocrinopathies) (see Chapter 7 ).
Lungs
- •
Hypoxia secondary to pulmonary disease results in secondary polycythemia.
- •
Idiopathic pulmonary hemosiderosis is a chronic disease characterized by recurrent intra-alveolar microhemorrhages with pulmonary dysfunction, hemoptysis, and hemosiderin-laden macrophages, resulting in iron-deficiency anemia. A precise diagnosis can be established by the presence of siderophages in the gastric aspirate. Apart from a primary idiopathic type, there is also a variant associated with hypersensitivity to cows’ milk and one that occurs with a progressive glomerulonephritis (Goodpasture syndrome). Treatment is controversial and may involve:
- •
Corticosteroids
- •
Withdrawal of cow’s milk
- •
Use of oral or IV iron when indicated
- •
Packed red cell transfusions when indicated.
- •
Skin
Mast Cell Disease
Mast cell disease or mastocytosis is associated with an abnormal accumulation of mastocytes (more closely related to monocytes or macrophages rather than to basophils) in the dermis (cutaneous mastocytosis) or in an internal organ (systemic mastocytosis). The systemic form is rare in children. In children, this condition is more common under 2 years of age. It usually presents either as a solitary cutaneous mastocytoma or, more commonly, as urticaria pigmentosa. Involvement beyond the skin is unusual in children, but splenomegaly and bone lesions have been reported. No reports of bone marrow disease in either acquired or congenital mastocytosis have been reported.
Eczema and Psoriasis
Patients with extensive eczema and psoriasis commonly have anemia. The anemia is usually normochromic and normocytic (anemia of chronic disease) and mild in most cases, but severely affected individuals can have hemoglobin levels less than 9 g/dl.
Dermatitis Herpetiformis
- •
Macrocytic anemia secondary to malabsorption.
- •
Hyposplenism: Howell–Jolly bodies may be present on blood smear.
Dyskeratosis Congenita
This disease is characterized by ectodermal dysplasia and aplastic anemia (see Chapter 8 ). The aplastic anemia is associated with high MCV, thrombocytopenia, and elevated fetal hemoglobin. This may occur before the onset of skin manifestations.
Hereditary Hemorrhagic Telangiectasia
This autosomal dominant disorder is associated with a bleeding disorder. Easy bruisability, epistaxis, and respiratory and gastrointestinal bleeding may be caused by telangiectatic lesions.
Ehlers–Danlos Syndrome
This condition may be associated with platelet dysfunction: reduced aggregation with ADP, epinephrine, and collagen. An unusual sensitivity to aspirin is described in type IV Ehlers–Danlos syndrome (see Chapter 14 ).
Chronic Illness
Chronic illnesses such as cancer, IBD, connective tissue disease, and chronic infection are associated with anemia. The anemia has the following characteristics:
- •
Normochromic, normocytic, occasionally microcytic
- •
Usually mild, characterized by decreased plasma iron and normal or increased reticuloendothelial iron
- •
Impaired flow of iron from reticuloendothelial cells to the bone marrow
- •
Decreased sideroblasts in the bone marrow.
The tests to differentiate the anemia of chronic illness from iron-deficiency anemia are listed in Table 2.4 and therapeutic options for the treatment of anemia in chronic disease are outlined in Table 2.5 .
| Variable (serum levels) | Anemia of chronic disease | Iron-deficiency anemia | Both conditions b |
|---|---|---|---|
| Iron | Reduced | Reduced | Reduced |
| Transferrin | Reduced to normal | Increased | Reduced |
| Transferrin saturation | Normal to mildly reduced | Reduced | Reduced |
| Ferritin | Normal to increased | Reduced | Reduced to normal |
| Soluble transferrin receptor | Normal | Increased | Normal to increased |
| Cytokine levels | Increased | Normal | Increased |
a Relative changes are given in relation to the respective normal values.
b Patients with both conditions include those with anemia of chronic disease and true iron deficiency.
| Treatment | Anemia of chronic disease | Anemia of chronic disease with true iron deficiency |
|---|---|---|
| Treatment of underlying disease | Yes | Yes |
| Transfusions a | Yes | Yes |
| Iron supplementation | No b | Yes c |
| Erythropoietin agents | Yes | Yes, in patients who do not have a response to iron therapy |
a This treatment is for the short-term correction of severe or life-threatening anemia. Potentially adverse immunomodulatory effects of blood transfusions are controversial.
b Although iron therapy is indicated for the correction of anemia of chronic disease in association with absolute iron deficiency, no data from prospective studies are available on the effects of iron therapy on the course of underlying chronic disease.
c Overcorrection of anemia (hemoglobin >12 g/dl) may be potentially harmful to patients; the clinical significance of erythropoietin-receptor expression on certain tumor cells needs to be investigated.
In inflammatory diseases, cytokines released by activated leukocytes and other cells exert multiple effects that contribute to the reduction in hemoglobin levels. The pathophysiology of anemia of chronic disease is shown in Figure 2.2 :
- 1.
Interleukins (IL), especially IL-6 along with endotoxin, induce hepcidin synthesis in the liver. Hepcidin in turn binds to Ferroportin located both in the GI tract and the reticuloendothelial system. Hepcidin binding induces the degradation of Ferroportin, sequestering oral intake iron from the GI tract and reticuloendothelial iron from storage sites. Hence in the classic patient with the anemia of chronic illness, the serum iron will be low, but there will also be a low level of transferrin iron-binding capacity secondary to a suppression of protein synthesis. This leads to a normal, or slightly diminished, iron saturation. The serum ferritin is then paradoxically elevated secondary to the hepcidin-induced sequester. The adult literature recognizes gray zone cases with normal or lower iron saturations and a low normal ferritin. In these situations laboratory testing for soluble transferrin receptor (which is elevated directly in response to iron deficiency) is used to direct iron treatment. These patients might also be simply given an intravenous iron challenge which will demonstrate improvement over 7–10 days if iron deficiency is present.
- 2.
Inhibition of erythropoietin release from the kidney (especially by IL-1β and tumor necrosis factor-alpha (TNF-α)) leads to reduced erythropoietin-stimulated hematopoietic proliferation.
- 3.
Direct inhibition of the proliferation of erythroid progenitors (especially by TNF-α, interferon-gamma (IFN-γ) and IL-1β).
- 4.
Increased erythrophagocytosis by reticuloendothelial macrophages.

Treatment involves treating the underlying illness.
Inflammatory Bowel Disease as a Model for Anemia of Chronic Illness
- •
In both Crohn’s disease and ulcerative colitis the anemia of chronic illness is often seen—sometimes before gastrointestinal symptomatology manifests.
- •
It is often associated with concomitant iron deficiency due to bleeding from the involved bowel.
- •
The patient may present with mild normochromic anemia or severe microcytic anemia.
In an older child or adolescent presenting with iron deficiency a detailed history of gastrointestinal symptoms must be pursued and any suggestion of anemia of chronic illness may alter the type and route of iron medication. If a patient with IBD presents with anemia, iron saturation and ferritin should be assessed prior to the initiation of treatment. The anemia of chronic illness is not iron-deficient erythropoiesis. It is a balance between the effect of elevated hepcidin in sequestering iron and a direct effect of cytokines slowing down erythropoiesis—and hence diminishing the erythropoietic call for iron. If the patient has a very low iron saturation and an elevated ferritin, oral iron is likely to have less effect given the mucosal block to iron in the anemia of chronic illness. Intravenous iron preparations will bypass that block and also diminish the additional gastrointestinal toxicity of oral iron. The administration of iron alone may not ameliorate the situation as patients may have such a severe effect of inflammation on erythropoiesis that they may require simultaneous administration of erythropoietin in pharmacologic doses along with iron. These considerations are identical to those faced in other conditions with major ongoing inflammation (e.g., juvenile rheumatoid arthritis).
Connective Tissue Diseases
Rheumatoid Arthritis
- •
Anemia of chronic illness (normocytic, normochromic)
- •
High incidence of iron deficiency
- •
Leukocytosis and neutropenia common in exacerbations of juvenile rheumatoid arthritis (JRA)
- •
Thrombocytosis associated with a high level of IL-6 occurs in many patients, although there may be transient episodes of thrombocytopenia.
Felty Syndrome
- •
Triad of rheumatoid arthritis, splenomegaly, and neutropenia.
- •
Patients may be at risk for life-threatening bacteremia. Splenic dysfunction resulting in infection with encapsulated organisms has been observed.
- •
Treatment involves controlling the rheumatoid arthritis, which often leads to improvement in the anemia. Parenteral antibiotics, with coverage for encapsulated organisms, for febrile episodes is recommended.
- •
G-CSF may be used in urgent situations although but there are concerns about splenic rupture with the use of this G-CSF because of case reports of spontaneous rupture of the spleen in Felty syndrome.
Systemic Lupus Erythematous
- •
Two types of anemia are common: anemia of chronic illness (normocytic, normochromic) and acquired, autoimmune direct antiglobulin (DAT)-positive hemolytic anemia.
- •
Neutropenia is common as a result of decreased marrow production and immune-mediated destruction.
- •
Lymphopenia with abnormalities of T-cell function.
- •
Immune thrombocytopenia: Antiphospholipid antibodies may be present which prolong the aPTT but are associated with severe thrombosis (lupus anticoagulant).
Polyarteritis Nodosa
- •
Microangiopathic hemolytic anemia may be associated with renal disease or hypertensive crises.
- •
Prominent eosinophilia.
Wegener Granulomatosis
This autoimmune disorder is rare in children. Hematological features include:
- •
Anemia: normocytic; RBC fragmentation with microangiopathic hemolytic anemia
- •
Leukocytosis with neutrophilia
- •
Eosinophilia
- •
Thrombocytosis.
Kawasaki Syndrome
This syndrome is characterized by:
- •
Mild normochromic, normocytic anemia with reticulocytopenia
- •
Leukocytosis with neutrophilia and toxic granulation of neutrophils and vacuoles
- •
Decreased T-suppressor cells
- •
High C 3 levels
- •
Increased cytokines IL-1, IL-6, IL-8, interferon-α, and TNF
- •
Marked thrombocytosis (mean platelet count 700,000/mm 3 )
- •
DIC.
Henoch–Schönlein Purpura
Henoch–Schönlein purpura (HSP), also called anaphylactoid purpura, is associated with systemic vasculitis characterized by unique palpable, erythema multiforma-like purpuric lesions, transient arthralgias or arthritis (especially affecting knees and ankles), colicky abdominal pain, and nephritis.
- •
Anemia occasionally occurs as a result of GI bleeding or decreased RBC production caused by renal failure.
- •
Transient decreased Factor XIII activity may occur, which may play a role in either gastrointestinal bleeding or HSP.
- •
Vitamin K deficiency from severe vasculitis-induced intestinal malabsorption has been reported.
Infections
Anemia
- •
Chronic infection is associated with the anemia of chronic illness.
- •
Acute infection, particularly viral infection, can produce transient bone marrow aplasia or selective transient erythrocytopenia.
- •
Parvovirus B19, with tropism for the developing red cell, infection in patients with an underlying hemolytic disorder (such as sickle cell disease, hereditary spherocytosis) can produce a rapid fall in hemoglobin and an erythroblastopenic crisis marked by anemia and reticulocytopenia. There may be an associated neutropenia and less commonly, thrombocytopenia.
- •
Many viral and bacterial illnesses may be associated with hemolysis.
White Cell Alterations
- •
Viral infections can produce leukopenia and neutropenia. Neutrophilia with an increased band count and left shift frequently results from bacterial infection.
- •
Neonates, particularly premature infants, may not develop an increase in white cell count in response to infection. Neonatal neutropenia may be serious and requires investigation and treatment. G-CSF has been used and found to be helpful in randomized clinical trials.
- •
Eosinophilia may develop in response to parasitic infections.
Clotting Abnormalities
Severe infections, for example Gram-negative sepsis, can produce DIC. Polymicrobial sepsis (including both aerobic and anaerobic organisms) in the head and neck region may cause thrombosis of major vessels. When this occurs in the jugular veins it leads to a constellation of findings called Lemierre’s syndrome (suppurative thrombophlebitis with inflammation starting in the pharynx and spreading to the lateral parapharyngeal tissues in association with jugular vein thrombosis).
Thrombocytopenia
Infection can produce thrombocytopenia through decreased marrow production, immune destruction, or DIC.
Viral and Bacterial Illnesses Associated with Marked Hematologic Sequelae
Parvovirus
Parvovirus B19 has a peculiar predilection for red cell precursors in the bone marrow. It has preference for the red cell precursors because it uses P antigen as a receptor. This viral infection is associated with a transient erythroblastopenic crisis, particularly in individuals with an underlying hemolytic disorder. In addition, it can produce thrombocytopenia, neutropenia, and a hemophagocytic syndrome. In immunocompromised individuals, parvovirus B19 infection can produce prolonged aplasia. Bone marrow aspirate shows decreased or arrested maturation of erythroid precursors and the pathognomonic “giant pronormoblasts.”
Epstein–Barr Virus
Epstein–Barr Virus (EBV) infection is associated with the following hematologic manifestations:
- •
Atypical lymphocytosis
- •
Acquired immune hemolytic anemia
- •
Agranulocytosis
- •
Aplastic anemia, rarely
- •
Lymphadenopathy and splenomegaly
- •
Immune thrombocytopenia.
EBV infection also has immunologic and oncologic associations (see Chapter 16 ). Some of the EBV-associated lymphoproliferative disorders are given in Table 2.6 .
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


