Chapter Outline
NEUROLOGIC AND PSYCHIATRIC DISORDERS
Thrombocytopenia and Pregnancy
Disseminated Intravascular Coagulation and Pregnancy
Sickle Cell Disease and Pregnancy
Inherited Bleeding Disorders and Pregnancy
Inherited Hypercoagulable States and Pregnancy
* This chapter is modified from the original chapter written by Dr. R. Alan B. Ezekowitz and includes material written by Dr. Samuel E. Lux, IV, with his permission.
Evaluation of almost any patient who seeks medical attention, particularly in a hospital setting, includes the obligatory complete blood cell count. The vast majority of these patients do not have a primary hematologic problem. However, there are many pathophysiologic paradigms that have secondary effects on hematopoiesis and/or the coagulation system. One of the most common examples of these effects includes the anemia of chronic disease or inflammation, which is associated with a wide variety of acute and chronic illnesses, including cancer, collagen vascular disease, severe tissue injuries, renal failure, and infectious processes. A detailed discussion of the anemia of chronic disease can be found in Chapter 11 .In this chapter, both an organ system and disease-specific approach is used. The goal of this chapter on the hematologic manifestations of systemic diseases is to focus on information that is not included in other sections of this book. For overlapping topics, the reader will be guided to other relevant chapters throughout the book.
Collagen Disorders
Collagen Vascular Disease
Many of the hematologic manifestations of the various collagen vascular diseases are similar. However, the characteristics that are unique to each syndrome are emphasized in this section.
Systemic Lupus Erythematosus
Anemia.
Anemia is the most common hematologic abnormality in systemic lupus erythematosus (SLE). There are multiple causes of anemia in this disease, although the anemia of chronic disease is probably the most common (see Chapter 11 ).
Acquired autoimmune hemolytic anemia (AIHA) may precede the onset of active SLE. It is typically characterized by a warm-type AIHA with a serologic pattern of immunoglobulin (Ig) G plus complement on the red blood cell surface. Although positive results on an antiglobulin test are common, true hemolysis is seen in less than 10% of patients. Chapter 13 details the pathophysiology and management of AIHA.
Mechanical hemolytic anemia may be seen in SLE-associated thrombotic thrombocytopenic purpura (TTP). Myelofibrosis has also been described in several patients with SLE; immune complexes have been suggested but not proven to be the cause of this rare complication of SLE. Aplastic anemia has been reported in association with SLE.
Leukopenia.
A reduction in the total white blood cell count is seen in most patients but is more common in adults than children. The leukopenia is usually caused by a combination of decreased numbers of granulocytes and lymphocytes. Lymphopenia may be severe in SLE and is sometimes due to IgG or IgM antilymphocyte antibodies. The magnitude of the reduction in T cells parallels disease activity and is associated with defects in cellular immunity, as measured by skin tests of delayed hypersensitivity, blast transformation in response to mitogen (phytohemagglutinin and concanavalin A), and macrophage-inhibiting factor production. Reduced numbers of regulatory T cells contribute to the loss of T- and B-cell tolerance in SLE. Unlike T-cell function, B-cell function is increased despite the decrease in the absolute number of B cells with emergence of autoreactive B cells. There is actually an increase in IgG-synthesizing peripheral blood lymphocytes, as well as elevated numbers of cells capable of binding native DNA and elevated numbers of IgM- and IgG-producing cells with antibody specificity against DNA antigen. These changes in peripheral blood lymphocytes are associated with certain morphologic alterations. A strongly basophilic cytoplasm and a high nuclear-to-cytoplasmic ratio are noted in many lymphocytes believed to be “immunoblasts.” On electron microscopic examination, inclusions are identified within lymphocytes that appear as undulating tubules characteristically associated with the endoplasmic reticulum.
Antibodies to granulocytes have been proposed as one mechanism for the granulocytopenia. Granulocyte antibodies and peripheral granulocyte destruction have been observed. Bone marrow depression of granulocyte formation may also occur. Sera from patients with SLE can inhibit mouse bone marrow colony-forming units. Qualitative abnormalities in granulocyte function have also been noted. Although several investigators have found no abnormalities in chemotaxis, others have observed abnormal migration by the skin window technique and diminished in vitro phagocytosis. These defects parallel the observed depressions in complement. The qualitative defects in granulocyte function may be the result of altered humoral factors rather than defects in the phagocytic cells themselves.
Thrombocytopenia.
Thrombocytopenia secondary to platelet immune sensitization is the most common immune-mediated hematologic manifestation of SLE (see Chapter 34 ). Thrombocytopenia may be an initial manifestation of SLE, and consideration of SLE should be part of the evaluation of immune thrombocytopenia (ITP) in adults and adolescent females. Occasionally, ITP may manifest years before serologic evidence or clinical manifestations of SLE are detectable. Similar to primary ITP, platelet survival is shortened and there are typically increased numbers of bone marrow megakaryocytes. The platelet antibody is commonly an IgG with a molecular mass of 150,000 to 330,000 daltons, and it binds complement. ITP in patients with SLE has the same therapeutic implication as primary ITP, that is, corticosteroids or intravenous immune globulin as primary intervention, with other treatments, including rituximab and splenectomy, reserved for patients who fail these approaches.
Rarely, a qualitative defect in platelet function may also be present with an antibody that interferes with platelet aggregation. Thrombocytopenia may be seen in association with SLE and TTP.
Lupus Anticoagulant and Antiphospholipid Syndrome.
Patients with SLE may also have a circulating anticoagulant in their plasma. The antibody-anticoagulant is directed against various phospholipids involved in phospholipid-dependent anticoagulation assays. Most investigators have reported that lupus anticoagulant is an immunoglobulin that may be of the IgG, IgM, or mixed IgG-IgM class. The pathologic significance of the lupus anticoagulant and other antiphospholipid antibodies is their association with thrombosis, mainly venous, as well as miscarriages and spontaneous abortions (see Chapter 35 ). Among adult SLE patients, 12% to 30% have anticardiolipin antibodies and 15% to 34% have lupus anticoagulant antibodies. The prevalence of thrombosis in pediatric SLE patients with lupus anticoagulant is not known. In adults, the prevalence of thrombosis is about 30%, with the predominant site being the legs in 66%, followed by the peripheral arteries in 10% and the cerebral arteries in 2%.
The partial thromboplastin time is invariably prolonged in patients with the lupus anticoagulant and is considered to be the most sensitive screening test. The inhibitor is suspected when the addition of normal plasma to the patient’s plasma fails to correct the defect. The prothrombin time in patients with the lupus anticoagulant is often normal or minimally prolonged because of the influence of the inhibitor. True prothrombin deficiency may occur in patients with the inhibitor, but it is rare. Prolonged thrombin times in the absence of elevated levels of fibrin split products may occasionally be seen. The exact significance of this finding is unclear.
A minority of patients have a catastrophic form of the disease. This often fatal syndrome is defined by the clinical involvement of at least three different organs over a period of days to weeks, with pathologic evidence of multiple occlusions of large or small vessels. In general, involvement of small vessels is more common in these patients, who tend to present with an acute thrombotic microangiopathy involving the kidney, lungs, central nervous system, heart, or skin, along with disseminated intravascular coagulation (DIC). The mortality rate is 30% to 50%. Aggressive therapy with combinations of anticoagulants, corticosteroids, intravenous immune globulin, and plasmapheresis have been tried in anecdotal reports with varying success.
In children, the lupus anticoagulant is usually found coincidentally on routine preoperative coagulation screening. In the majority of cases, this lupus anticoagulant is not associated with SLE or bleeding symptoms. In these cases, the lupus anticoagulant typically resolves spontaneously within weeks to months.
Juvenile Idiopathic Arthritis (Rheumatoid Arthritis)
Anemia in rheumatoid arthritis is caused typically by both anemia of chronic disease and iron-deficiency anemia. A high incidence of iron deficiency has historically been seen in children with juvenile idiopathic arthritis (JIA), because of gastrointestinal blood loss secondary to long-standing use of aspirin and nonsteroidal analgesics in the symptomatic treatment of this disease. The severe inflammatory nature of this disorder makes it a model for anemia of chronic disease, a sideropenic anemia with reticuloendothelial iron overload. Iron is not only loaded inappropriately into the reticuloendothelial system but also accumulates in the involved joint, possibly playing a role in joint destruction.
Microcytosis is very common in juvenile chronic forms of arthritis, with a rate of occurrence of about 40%. The degree of microcytosis relates directly to disease activity. Occasionally, macrocytic anemia develops in patients with JIA. Although abnormal vitamin B 12 metabolism has been suggested as a cause, this is not seen in children. Folate metabolism may, however, be abnormal. Some children with rheumatoid arthritis have diminished plasma and red blood cell folate levels. Increased folic acid plasma clearance and reduced protein binding have also been observed.
A few patients have had mild shortening of red blood cell survival as a result of an extracorpuscular defect. More severe hemolytic anemia is much less common than in other collagen vascular diseases. Erythroid aplasia responsive to corticosteroid therapy has been reported in a child with juvenile rheumatoid arthritis. Circulating inhibitors of erythropoiesis have also been noted.
Leukocytosis and neutrophilia are common during acute flare-ups of JIA; however, this is uncommon in the adult form of the disorder. Neutrophil chemotaxis may be mildly diminished. Phagocytosis is normal or mildly impaired. Nitroblue tetrazolium dye reduction may be increased during active phases of this disease. These minimal alterations in white blood cell function do not produce any clinical disturbances. Wound healing, for example, is normal even after surgery in patients who have not been receiving high-dose corticosteroid therapy. A peripheral blood eosinophil count greater than 5% has been noted in slightly more than 50% of children with JIA. Some will also demonstrate basophilia and plasmacytoid lymphocytes.
Secondary thrombocytosis is common in JIA, as part of the inflammatory response. A consumptive coagulopathy may be seen in association with systemic JIA. Acquired circulating inhibitor to factor VIII has been described in JIA and may cause bleeding. However, clinical problems related to coagulation abnormalities in this disorder are rare.
Felty Syndrome
Felty originally described the triad of rheumatoid arthritis, splenomegaly, and neutropenia in 1924. Whether Felty syndrome occurs with JIA is unclear. Splenomegaly is seen in about 20% of all children with JIA, especially in those with the acute extraarticular exacerbations of this disease. However, splenomegaly alone does not suggest Felty syndrome.
The etiology for the neutropenia of Felty syndrome is multifactorial. The neutropenia is not always a consequence of hypersplenism per se because only 60% of adults who undergo splenectomy experience resolution of their neutropenia. In 50% of patients, a maturation arrest bone marrow pattern is demonstrated, possibly from immune humoral or cellular mechanisms. Neutropenia in association with cytotoxic lymphocytes directed against granulocyte progenitors has also been reported, as have lower than normal levels of granulocyte colony-stimulating factor (G-CSF) activity. The neutropenia of Felty syndrome may also be immunologic in origin, because some patients have IgG antibodies directed against neutrophils.
The neutropenia may be associated with serious infections. Recombinant human G-CSF and granulocyte-macrophage colony-stimulating factor (GM-CSF) both effectively raise the neutrophil count in essentially all patients with Felty syndrome. The neutrophil count decreases again once the growth factor treatment is stopped, but often it stabilizes at a level higher than it was before treatment. The drug can be used long term and may be cost-effective in selected patients with a high incidence of infections and repeated hospitalizations. Methotrexate treatment in Felty syndrome can be effective in correcting the neutropenia. Splenectomy may be indicated in those in whom G-CSF or GM-CSF is ineffective or rapid blood cell improvement is desired. A rise in the neutrophil count after splenectomy is experienced by 50% to 80% of patients.
Vasculitis
Polyarteritis Nodosa
Microangiopathic hemolytic anemia can be associated with renal disease or hypertensive crises in polyarteritis nodosa. The hemolytic process in these circumstances parallels disease activity. Anemia of chronic disease is common in polyarteritis nodosa.
Neutropenia occurs and is associated with the presence of antineutrophil cytoplasmic antibodies. Neutrophilia and eosinophilia are also common. The eosinophilia may be marked, particularly in those patients with clinically apparent pulmonary involvement.
Wegener Granulomatosis
Wegener granulomatosis, characterized by necrotizing vasculitis (particularly in the lungs and kidneys), is rare but can affect children. The disease can also occur in the neonatal period. The course of the disease is marked by fever, cough, hemoptysis, epistaxis, nasal discharge, obliteration of the nasal sinuses, and nodular pulmonary infiltrates. Renal failure may occur. A microangiopathic hemolytic anemia can develop. An associated anemia of chronic disease is common.
The white blood cell count is elevated, often coupled with an eosinophilia. The disease is characterized by marked thrombocytosis. Autoantibodies directed against cytoplasmic components of neutrophil granulocytes and monocytes have been described as a disease-specific marker for Wegener granulomatosis.
Connective Tissue Disorders
Bleeding and bruising are common manifestations of connective tissue disorders related to fragility of capillaries and perivascular connective tissue rather than disorders of clotting or platelet function. Primary hemostasis depends on the interaction of the vessel wall, platelets, and the clotting cascade. In connective tissue disorders, the connective tissues of the skin, subcutaneous tissue, and blood vessel wall have increased fragility, which portends increased bruising and bleeding. Laboratory evaluation for disorders of platelet aggregation and clotting factors is typically normal. Genetic counseling is recommended when a connective tissue disorder is suspected.
Ehlers-Danlos Syndrome
Most Ehlers-Danlos subtypes are caused by mutations in fibrillar collagens. The elastic tissue defects result in an increased bleeding tendency. Different forms of Ehlers-Danlos have been associated with platelet and coagulation defects, including disorders of platelet aggregation and factor deficiencies. It is not clear whether these hematologic defects are sporadic associations in individual patients or true associations with the subtypes of Ehlers-Danlos.
In Ehlers-Danlos syndrome, the bleeding symptoms are typically easy bruising, gingival bleeding, prolonged bleeding with surgical or dental procedures, and menorrhagia. Other common clinical findings include skin hyperextensibility, “cigarette paper–like” scars, and joint hypermobility. The vascular Ehlers-Danlos syndrome, type IV, is an autosomal dominant disorder caused by mutations in the collagen type III alpha 1 (COL3A1) gene. Type IV Ehlers-Danlos is associated with small and large vessel bleeding, including arterial bleeding, and spontaneous ruptures of the bowel, uterus, and lungs. Nonemergent surgical interventions are to be avoided in these individuals owing to the high risk for perisurgical bleeding and complications. With serious bleeding in Ehlers-Danlos syndrome patients, efficacy of desmopressin and of factor VIIa has been reported.
Other Inherited Connective Tissue Disorders
Marfan syndrome is not associated with fragility of capillaries or small to medium-sized arteries or veins. However, many Marfan syndrome patients report easy bruising, which has not been explained. A defective fibronectin has been proposed as the cause of the hypermobility and platelet dysfunction in some patients. Easy bruising has also been reported in individuals with osteogenesis imperfecta.
Scurvy
Ascorbic acid functions as a cofactor, enzyme complement, cosubstrate, and antioxidant for a wide variety of metabolic processes. Vitamin C is involved in reactions involving iron, copper, vitamin E, and folic acid. Deficient intake of ascorbic acid results in impaired collagen synthesis, mitochondrial fatty acid transport, and synthesis of neurotransmitters. Although scurvy is uncommon in developed countries, certain patient subpopulations remain at risk for this type of nutritional deficiency, such as those with neurologic impairment, psychiatric disease, alcoholism, small bowel disease, and iron-overload disorders associated with vitamin C renal wasting. Scurvy occurs when vitamin C has been eliminated from the diet for at least 3 months. Hematologic symptoms of scurvy arise mainly from defective collagen synthesis and include gingival bleeding, subperiosteal hemorrhage with painful joint bleeding, and organ hemorrhage. Scurvy is associated with a multifactorial anemia related to bleeding, dietary deficiencies, oxidative hemolysis, and altered metabolism of iron and folate. Other manifestations include poor wound healing, defective dentine formation, tooth and hair loss, and bony changes. The symptoms of scurvy, including the bleeding manifestations, rapidly respond to enteral vitamin C supplementation.
Infectious Diseases
General Hematologic Signs of Infection
Red Blood Cell Disturbances
The anemia of chronic disease is common to all infections. Even common childhood infections, especially those associated with inflammation, will cause a decline in hemoglobin concentration. During active inflammation, the hemoglobin concentration declines about 13%, usually within 1 week, followed by an increase of nearly 25% during resolution of the active inflammation. The actions of hepcidin probably deprive microorganisms of iron, which is an essential nutrient for microbial replication, and may be a component of host immunity against infection. Serum levels of iron decrease after a bacterial challenge, and the lactoferrin released from stimulated granulocytes is a conduit for this iron to return to reticuloendothelial cells. Many examples of bacterial pathogenicity being dependent on iron availability (e.g., Neisseria gonorrhoeae, Neisseria meningitidis ) support the hypothesis that the anemia of infection is the body’s compromise in an attempt to deprive invading microorganisms of iron.
Although some infections, particularly viral ones, such as parvovirus, cause transient bone marrow aplasia or selective erythroid aplasia, anemia from this cause is rare because of the long life span of red blood cells. In contrast, patients with chronic hemolytic anemias may experience a rapid decrease in hemoglobin concentration or an “aplastic” crisis during viral and some bacterial infections.
Severe hemolytic anemia may be observed in certain types of infections. Clostridial infections may result in a high titer of hemolysins and cause severe anemia. A similar severe anemia may result from sepsis related to other bacterial organisms, including staphylococci, streptococci, pneumococci, and Haemophilus influenzae. Immune hemolytic anemia mediated by a cold agglutinin may be observed with Listeria and Mycoplasma infections and occasionally with infections by other organisms such as Epstein-Barr virus.
Many viral illnesses may be associated with what appears to be a mild hemolytic anemia for which no pathologic mechanism has been defined. The most common morphologic finding in these circumstances is poikilocytosis. Certain viruses, such as most strains of influenza virus, contain neuraminidase activity, which is, at least theoretically, capable of affecting the sialic acid content of the red blood cell membrane. Whether this plays any significant role in the hemolysis associated with some viral diseases is not known.
Many congenital infections, including cytomegalovirus, herpes simplex, rubella, toxoplasmosis, and syphilis, produce profound hemolytic anemia in the neonatal period, even though these same agents may not significantly alter red blood cell survival at other times of life.
White Blood Cell Disturbances
The white blood cell count may be normal, low, or high with infection. Viral illnesses may be associated with leukocyte counts lower than 5000 cells/mm 3 , although bacterial diseases of certain types or overwhelming sepsis of any type may also cause leukopenia. The most common viral illnesses associated with leukopenia are infectious hepatitis, infectious mononucleosis, rubella, measles, and, occasionally, influenza. Of the bacterial infections, shigellosis may produce leukopenia with a marked increase in band cell forms. Sepsis caused by meningococci, pneumococci, staphylococci, and a few other bacterial pathogens may also cause leukopenia.
Neutrophilia, with or without an increase in band cell count, is a common result of bacterial infection. White blood cell and neutrophil counts do not differ between children of different races with bacteremia. Occasionally, viral illness also initially will be manifested as neutrophilia. A variety of morphologic changes may appear in the neutrophils of patients with infection. Döhle bodies ( Fig. 37-1 ), which are pale blue cystlike inclusion bodies usually located in the periphery of the cytoplasm of neutrophils, may appear in bacterial infections. They are occasionally associated with viral illness but are also commonly seen in patients with burns, massive trauma, and cancer, as well as in pregnancy and after the use of cyclophosphamide. In addition, Döhle bodies are seen in the May-Hegglin anomaly. Increased size of neutrophil granules (“toxic granulation”) may be found in both bacterial and viral illnesses, as well as in many of the other disorders associated with the presence of Döhle bodies. Vacuolization of the cytoplasm of neutrophils is the next most common morphologic abnormality of neutrophils in patients with significant bacteremia. In overwhelming sepsis, the organisms are frequently evident in the vacuoles of the polymorphonuclear cells. In a study of the neutrophils of patients with bacteremia, Zipursky and associates found toxic granulation, Döhle bodies, and vacuolization in 75%, 29%, and 24%, respectively, of the patients studied.
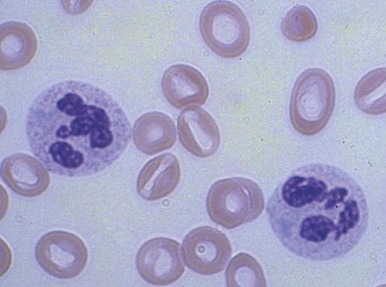
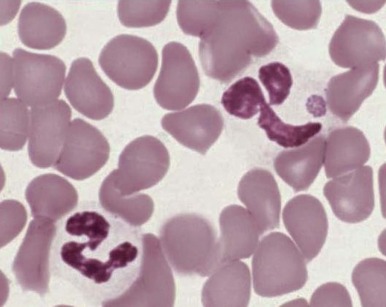
Increased neutrophil alkaline phosphatase activity and nitroblue tetrazolium dye reduction may also occur, but neither of these characteristics is specific for bacterial infection. Infections may be associated with development of the Pelger-Huët anomaly ( Fig. 37-2 ), in which granulocytes and eosinophils have one or two lobes per nucleus and assume a round, dumbbell, or peanut shape.
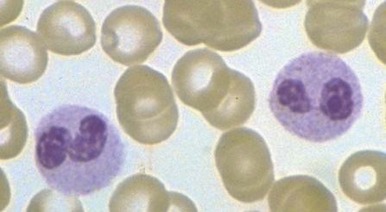
In neonates, especially those born prematurely, an increase in total white blood cell or mature neutrophil counts may not be seen in the presence of infection. In fact, a decrease in the neutrophil count often occurs. The most helpful signs of septicemia in this age group are an increase in the band cell count and the presence of toxic granulations and Döhle bodies.
Leukocytosis may result from lymphocytosis. The most common infections producing the greatest increases in lymphocyte counts are infectious mononucleosis, cat-scratch disease, and pertussis. Many other viral illnesses, such as cytomegalovirus, rubella, mumps, and hepatitis, may also cause an increase in the lymphocyte count. Lymphopenia of T lymphocytes is a common finding after measles infection.
Eosinophilia may reflect the presence of parasitic infections. In the United States, the most common cause of marked elevations in eosinophil counts is Toxocara infestation, which is often accompanied by high titers of isohemagglutinins. Other parasites commonly causing eosinophilia include organisms belonging to Trichinella, Echinococcus, Filaria, Strongyloides, Schistosoma, Enterobius, and Ancylostoma and tapeworms other than Echinococcus. Allergic sensitization to mites may cause eosinophilia, as well as fungal infections, especially aspergillosis. Eosinophilia is not specific for infestation. Marked degrees of eosinophilia may occur in association with prematurity. An absolute eosinophilia may be expected in about 75% of low-birth-weight infants. In some, the eosinophilia is marked (3000 cells/mm 3 ) and the maximal increase seems to occur at about the time that birth weight is regained, although this is not true in all infants.
Monocytosis is occasionally reported with specific infections, especially tuberculosis, syphilis, and subacute bacterial endocarditis. Monocytosis is often noted early in the course of many infections and again on recovery, particularly if it is associated with granulocytopenia.
Basophilia is rarely seen in infection but has been reported with tuberculosis, influenza, and hookworm infestation.
Clotting Abnormalities and Thrombocytopenia
DIC may be triggered by infectious processes. Of the infectious causes, gram-negative septicemia is probably most common. Meningococcus, Escherichia coli, Proteus, Pseudomonas, Aerobacter, and Klebsiella are among the most common etiologic agents recovered from the bloodstream. Gram-positive septicemia can cause a similar picture. The most common offender is Streptococcus pneumoniae, especially in asplenic individuals. Other gram-positive agents causing DIC include Staphylococcus aureus, other streptococcal species, and Clostridium. A wide range of viral infections may cause a consumptive coagulopathy that often leads to purpura fulminans. Among the most common agents are those that cause infectious hepatitis, measles, rubella, varicella, and infectious mononucleosis. Less common causes of DIC are severe mycoplasmal, rickettsial, and malarial infections.
Thrombocytopenia occurring separately from a true disseminated consumptive process is quite common in many infectious processes, especially in association with infectious mononucleosis, cytomegalovirus, rubella, measles, gram-negative bacteremia, and rickettsial diseases. Congenital viral infections and congenital syphilis and toxoplasmosis, if clinically apparent, are almost invariably associated with increased platelet turnover with or without thrombocytopenia. Pediatric immune thrombocytopenia is commonly associated with viral infections. Immune thrombocytopenia may also occur after immunization with the measles, mumps, and rubella vaccine. Corrigan found that thrombocytopenia without consumptive coagulopathy is an extremely common finding in infants and children with septicemia. In contrast, thrombocytosis is often present during the active phases of infectious processes. The platelet distribution width and mean platelet volume may be helpful in predicting whether the thrombocytopenia is due to infection. In the late neonatal period, thrombocytopenia associated with an infection dramatically increases the mean platelet volume and platelet distribution width as determined by electronic counting equipment.
Bone Marrow Abnormalities
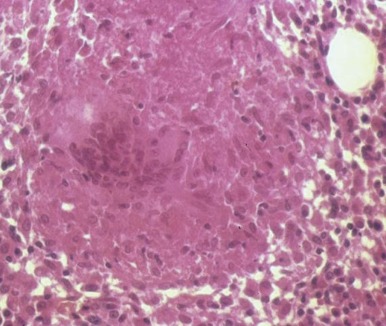
The bone marrow frequently contains clues as to the source of infectious diseases. Histoplasmosis ( Fig. 37-3 ), tuberculosis, kala-azar, Salmonella typhi, and Candida ( Fig. 37-4 ) all have been identified in marrow macrophages. Marrow granulomas may be a marker of disseminated tuberculosis or histoplasmosis ( Fig. 37-5 ). Histoplasmosis, tuberculosis, brucellosis, and Salmonella can be successfully cultured from the marrow aspirate.
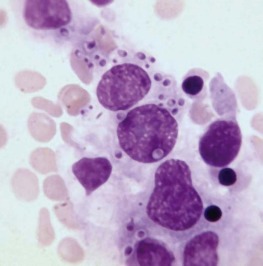
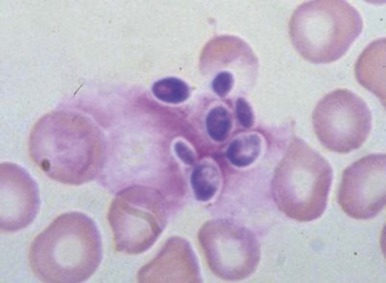
Hematologic Aspects of Specific Infections
Bacterial Infections
Clostridial Sepsis.
Clostridium perfringens septicemia is seen in a variety of clinical situations but must particularly be considered in patients with penetrating wounds, septic abortions, peritonitis after a perforated viscus, or cholecystitis or cholangitis; in immunosuppressed patients with gastrointestinal or hematologic malignancies; and in neonates with necrotizing enterocolitis. In patients with clostridial sepsis, severe, rapidly progressive intravascular hemolysis and microspherocytosis may occur ( Fig. 37-6 ). Hemolysis of the entire red blood cell mass has been reported. Complications include shock, acute renal failure, and death. Transfusion therapy may be ineffective. Antibiotics and hyperbaric oxygen occasionally have been used successfully to treat clostridial infections.
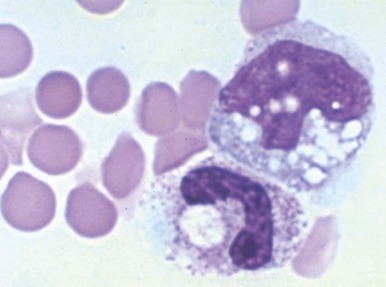
The mechanism of red blood cell damage is uncertain and may vary between patients. The bacteria produce several hemolytic toxins including α-toxin, a 43-kD protein that contains an NH 2 -terminal phospholipase domain and a COOH-terminal domain required for hemolysis, and θ-toxin, a 54-kD cholesterol-binding protein that aggregates and forms membrane pores leading to colloid osmotic hemolysis. α-Toxin appears to induce hemolysis through the activation of sphingomyelin metabolism. C. perfringens contains a neuraminidase that cleaves terminal sialic acids from red blood cell glycoproteins in some patients. The underlying galactose residues form the Thomsen-Friedenreich cryptoantigen (or T antigen). Anti-T antibodies are present in almost all adult plasma; thus T-antigen activation can lead to significant hemolysis. In infected infants and children who lack T antibodies, transfusion may lead to massive hemolysis and death. Rarely, T-antigen activation may precede the intravascular hemolysis, leading to early detection of clostridial sepsis and lifesaving therapeutic intervention.
Pertussis.
Pertussis may cause a marked increase in the white blood cell count, with elevations to 40,000 cells/mm 3 or higher, most of which are due to an increase in the lymphocyte count. Marked leukocytosis has been associated with a more severe course. Hyperleukocytosis with white blood cell counts greater than 100,000 cells/mm 3 has been associated with the presence of immature leukocytes and obstruction in the pulmonary microcirculation. Successful leukoreduction has led to clinical improvement in a number of young infants with severe pertussis.
Babesiosis.
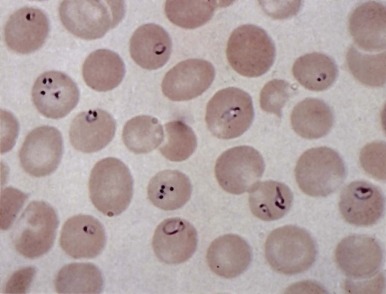
In healthy individuals, a mild hemolytic anemia is caused by Babesia microti and Babesia divergens microorganisms. Occasional cases of combined B. microti babesiosis and Lyme disease are described, attributable to the shared geographic locale of the tick vectors for these diseases. The disease can also be acquired by transfusion. Babesiosis is not life threatening except in asplenic subjects in whom the parasitized red blood cells are not contained. Then, the infection can be rapidly progressive and life threatening. In extreme cases, nearly all the red blood cells may be parasitized ( Fig. 37-7 ). Patients may have malaise, headache, fever, shaking chills, profuse sweating, jaundice, and dark urine. There may be intravascular hemolysis and, occasionally, pancytopenia and hemophagocytosis. Diagnosis is made by finding the parasites in blood smears or by serologic tests or amplification of parasitic DNA using the polymerase chain reaction (PCR) assay. Current treatment of symptomatic cases is quinine plus clindamycin, but treatment failures have been reported in asplenic patients. Exchange transfusion may reduce the parasite load and be beneficial.
Anaplasmosis (Ehrlichiosis).
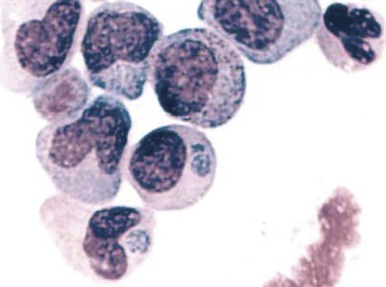
A variety of hematologic manifestations are found in infections with Ehrlichia chaffeensis, the causative bacteria of human monocytic ehrlichiosis (HME, Fig. 37-8 ), and Anaplasma phagocytophilum, the causative bacteria of human granulocytic anaplasmosis (HGA, Fig. 37-9 )). The disease is transmitted through tick bites, typically through the Lone Star tick or Ixodes scapularis, the shared tick vector of Lyme disease and babesiosis. Transmission has also been reported through blood transfusions. Symptoms are variable but are most severe in immunocompromised individuals. The obligate intracellular bacteria grow within membrane-bound vacuoles in leukocytes. General symptoms can include fever, headache, rash, and neurologic symptoms. Thrombocytopenia and leukopenia are common, often with both lymphopenia and neutropenia. Lymphopenia can be followed by an atypical lymphocytosis in the later stages of disease. Intraleukocytic morulae detected on review of the peripheral blood smear or buffy coat can be seen in up to 50% to 70% of infected individuals. Diagnosis can also be made through serologic tests or PCR assay. Doxycycline is the treatment of choice; however, rifampin has been reported to be effective in young patients.
Mycobacteria.
Tuberculosis produces a variety of hematologic abnormalities. Leukemoid reactions mimicking myeloproliferative disorders are common. Bone marrow involvement in miliary tuberculosis may result in a leukoerythroblastic pattern with teardrop-shaped red blood cells, nucleated red blood cells, and myeloblasts apparent on the peripheral blood smear. Bone marrow biopsy may show evidence of granulomas. Monocytosis is common, and thrombocytopenia and pancytopenia have been reported.
Viral Infections
Parvovirus B19.
Parvovirus B19, the cause of fifth disease (erythema infectiosum) in children, infects and kills early erythroid progenitors bearing the P blood group antigen. This infection, which is discussed in Chapter 6 , is not clinically significant unless the infection occurs in the setting of chronic hemolytic processes such as sickle cell disease or hereditary spherocytosis, or in patients with compromised immunity. In these situations, life-threatening red blood cell aplasia or even pancytopenia may be caused by this virus. The infection typically presents as fever, vomiting, and abdominal pain, along with pallor, fatigue, and other symptoms of anemia. Sometimes multiple family members are affected simultaneously. Parvovirus B19 infections are often associated with mild neutropenia or thrombocytopenia, and isolated cases of transient pancytopenia, hemophagocytosis, and transient myelodysplasia are reported. The virus is a particular danger to pregnant women because it can cause fetal death due to anemia and nonimmune hydrops fetalis.
Infectious Mononucleosis.
Infectious mononucleosis is caused by the Epstein-Barr virus (EBV) and historically associated with the triad of (1) the classic clinical picture, (2) atypical lymphocytosis, and (3) positive results on a heterophil antibody test. Serologic testing for EBV is now often used in addition or instead of the heterophil antibody test to confirm acute infection.
Epidemiology.
EBV infection is acquired at an early age in lower socioeconomic groups. In economically privileged children, infection is often delayed until adolescence and young adulthood. About 40% of American children are seropositive for EBV by 5 years of age. In a report from the U.S. Military Academy at West Point, 63.5% of entering cadets had EBV antibody, indicative of previous infection. During the college years, the infection rate among susceptible individuals is 12% to 15% per year.
The pattern of infection in different groups of susceptible persons is probably best explained by transmission of this virus in throat secretions. EBV is present in the saliva of most patients with infectious mononucleosis, in up to 20% of healthy persons with EBV antibodies, and in 50% or more of seropositive patients receiving immunosuppressive drugs. The ease with which EBV is recovered from the oral secretions of persons with primary or reactivated EBV infection suggests that a cell type that freely permits EBV replication exists in the oropharynx. The oropharyngeal epithelial cell may be the target cell type that is productively infected in infectious mononucleosis. Transmission requires intimate contact. Intrafamilial spread, however, does occur frequently, and, in this setting, an incubation period of 4 to 6 weeks has been demonstrated. Another possible route of transmission of EBV is transfusion through white blood cells.
Clinical Manifestations.
The classic clinical manifestations of infectious mononucleosis are generally seen only in adolescents and young adults. Younger children rarely exhibit the typical findings of this disease, and most generally have only a mild viral respiratory illness.
The onset of typical illness is usually a subtle prodrome consisting of fatigue, malaise, sweating, feverishness, and anorexia. Headache, nausea, and vomiting are seen frequently. The most common symptom during this period is a sore throat, which begins slowly and increases in intensity over a 1-week period. The usual findings of infectious mononucleosis then follow ( Table 37-1 ). The course of fever often follows a specific pattern, with no temperature elevation in the morning but daily afternoon or evening peaks of 38.3° C to 39.4° C (101° F to 103° F). Occasionally, higher temperatures are observed. The fever usually lasts about 2 weeks.
| Symptom or Sign | % |
|---|---|
| Adenopathy | 100 |
| Malaise and fatigue | 90-100 |
| Fever | 80-95 |
| Sweats | 80-95 |
| Sore throat, dysphagia | 80-95 |
| Pharyngitis | 65-85 |
| Anorexia | 50-80 |
| Nausea | 50-70 |
| Splenomegaly | 50-60 |
| Headache | 40-70 |
| Chills | 40-60 |
| Bradycardia | 35-50 |
| Cough | 30-50 |
| Periorbital edema | 25-40 |
| Palatal enanthema | 25-35 |
| Liver or splenic tenderness | 15-30 |
| Myalgia | 12-30 |
| Hepatomegaly | 15-25 |
| Rhinitis | 10-25 |
| Ocular muscle pain | 10-20 |
| Chest pain | 5-20 |
| Jaundice | 5-10 |
| Arthralgia | 5-10 |
| Diarrhea or soft stools | 5-10 |
| Photophobia | 5-10 |
| Rash | 3-6 |
| Conjunctivitis | 5 |
| Abdominal pain | 5 |
| Gingivitis | 3 |
| Pneumonitis | 3 |
| Epistaxis | 3 |
Lymphadenopathy is seen in all patients. Symmetric, moderately enlarged, discrete, slightly tender nodes, especially in the posterior cervical region, are most characteristic. Adenopathy is common in the axillary, epitrochlear, and inguinal areas. The nodes are not matted and do not show signs of heat, redness, or fluctuation. Rarely, enlargement of the mediastinal glands constitutes the only evidence of adenopathy and may be confused with a lymphomatous process.
Splenomegaly occurs in more than 50% of patients with infectious mononucleosis. The spleen is usually just barely palpable but on rare occasion may be quite large. It is smooth, soft to firm, and sometimes slightly tender. In some patients, splenomegaly persists for months, but the most common situation is resolution by 2 to 3 weeks after onset of the illness.
Hepatomegaly occurs in about 20% of patients, and clinical jaundice is seen in 10%. The jaundice is invariably mild. Despite the low rate of occurrence of apparent hepatic dysfunction, virtually all patients with infectious mononucleosis demonstrate abnormal liver enzyme levels. Except in rare patients with acute liver failure, the hepatitis associated with this illness is self-limited. There is no evidence that chronic liver disease or cirrhosis results from infectious mononucleosis.
Rashes occur occasionally, and their manifestation follows no particular pattern. The rash may be a diffuse, faint, erythematous or maculopapular eruption, or it may be urticarial, scarlatiniform, petechial, or herpetiform. In general, the rashes of infectious mononucleosis have no unique features and are of little or no help for diagnosis. Circulating immune complexes and complement sequence activation occur only when rashes are present. If ampicillin is administered to patients with infectious mononucleosis, a rash will develop in 69% to 100%. Other penicillins may cause this response but less often. This rash is not an allergic reaction, and these antibiotics may be administered subsequently without ill effect.
Only a few patients with infectious mononucleosis have no pharyngitis. Almost all demonstrate hyperplasia of the pharyngeal lymphoid follicles. The presence of an exudate is common, and membrane formation occurs frequently. The inflammation may be severe enough to cause respiratory obstruction. Peritonsillar abscesses may complicate the course of the illness. A palatal enanthema is seen in about a third of patients and consists of crops of sharply circumscribed petechiae, symmetrically distributed at the junction of the soft palate. Unfortunately, such petechiae are not specific for infectious mononucleosis and have been described in rubella and other viral disorders. In fact, the entire pharyngeal manifestation of infectious mononucleosis is clinically indistinguishable from that of streptococcal disease.
Periorbital edema is not rare and occasionally leads to the erroneous suspicion that renal disease or hypoproteinemia is present. The edema is self-limited and lasts only a few days.
Complications.
Much attention is paid to the complications associated with infectious mononucleosis, although their overall occurrence is low ( Table 37-2 ). Hematologic complications of infectious mononucleosis include disturbances resulting in anemia, granulocytopenia, thrombocytopenia, and occasional coagulation defects. The rate of occurrence of these hematologic abnormalities is shown in Table 37-3 .
| Type of Complication | Diagnosis or Description of Abnormalities |
|---|---|
| Neurologic | Bell palsy, cerebellar syndrome, encephalitis, encephalomyelitis, encephalomyelopathy, Guillain-Barré syndrome, meningitis, meningoencephalitis, myelitis, optic neuritis, peripheral neuritis, psychosis, radiculoneuritis, ataxia, positive Babinski sign, coma, convulsions, diplopia, extraocular palsy, facial diplegia, hemiplegia, hyperesthesia, meningismus, mental confusion, nystagmus, papilledema, psychotic reaction, ptosis, respiratory paralysis, positive Romberg sign, seizure, status epilepticus, scotomas |
| Cardiac | Electrocardiographic changes, myocarditis, pericarditis |
| Ocular | Conjunctivitis, diplopia, cyclic edema, hemianopia, lacrimal pericyclitis, nystagmus, optic neuritis, ptosis, retinal edema, retinal hemorrhage, retroorbital pain, scotomas, uveitis |
| Respiratory | Laryngeal obstruction, peritonsillar abscess, pharyngeal edema, pleural effusion, pleuritis, pneumonitis |
| Hematologic | Acquired hemolytic anemia, agranulocytosis, eosinophilia, fibrinolysis, pancytopenia, splenic rupture, thrombocytopenia |
| Digestive | Esophageal varices, gingivitis, hepatic dysfunction, hepatic necrosis, jaundice, melena |
| Renal | Hematuria, hemoglobinuria, nephritis, nephrotic syndrome, porphyrinuria, proteinuria |
| Other | Bullous myringitis, endocervicitis, orchitis, otitis media, pancreatitis, porphyria, rashes |
| Findings | % Positive |
|---|---|
| Lymphocytosis, relative and absolute | 100 |
| Atypical lymphocytes, definite * | 100 |
| Epstein-Barr virus antibody in serum | 100 |
| Heterophil antibody | 80-100 |
| Liver enzyme abnormalities | 80-100 |
| Leukocytosis | 60-80 |
| Neutropenia | 60-80 |
| Hyperbilirubinemia | 30-50 |
| Bone marrow granulomas | 50 |
| Slight thrombocytopenia | 25-50 |
| Increased cold agglutinins | 10-50 |
| Occult hemolysis | 20-40 |
| Hyperuricemia | 15-20 |
| Leukopenia | 10-20 |
| Severe thrombocytopenia with bleeding | Rare |
| Positive direct Coombs test results | Rare |
| Significant anemia (usually caused by hemolysis) | Rare |
* Twenty percent or more of white blood cells in peripheral blood.
AIHA can occur with EBV infection and developed in approximately 3% of West Point cadets with infectious mononucleosis. When hemolysis does occur, it usually begins 1 to 2 weeks into the course of the illness. The majority of occurrences terminate in less than 1 month, and chronic hemolysis is rare. Although usually mild, hemolysis occasionally occurs rapidly and can then result in severe anemia. Jenkins reported the first instance of hemolytic anemia in infectious mononucleosis that was mediated by the temporary induction of a high-thermal amplitude cold agglutinin of anti-i specificity. Since then, several series have verified the high incidence of anti-i antibody as the cause of immune-mediated hemolysis in infectious mononucleosis. It should be noted that although hemolysis is not common in infectious mononucleosis, the presence of anti-i is seen in as many as 50% of all patients. Not all instances of immune hemolysis in infectious mononucleosis are caused by anti-i antibodies. Anti-N antibodies have also been reported, and in some patients the nature of the antibody has not been identified.
Aplastic anemia has been reported to follow the onset of infectious mononucleosis. Aplastic anemia caused by EBV infection has also been reported after bone marrow transplantation. Localization of EBV in the bone marrow of some patients with aplastic anemia supports a causative role of the virus in these aplastic anemia patients.
Granulocytopenia is common during the acute phase of infectious mononucleosis. It is rarely severe but, on occasion, is associated with a secondary bacterial infection. Bone marrow myeloid hyperplasia with myeloid arrest is the most typical finding. Spontaneous resolution is the rule.
Immune thrombocytopenia occurs rarely in infectious mononucleosis. Most occurrences of thrombocytopenia are mild. The signs and symptoms of severe occurrences are similar to those of primary ITP. Severe hemorrhagic complications are rare. Treatment of the thrombocytopenia of infectious mononucleosis in children is guided by bleeding symptoms. Treatment approaches are similar to primary ITP with first-line therapies with corticosteroids and/or intravenous immune globulin.
The incidence of the neurologic complications reported varies from 0.37% to 7.3%, depending on the series. A rare occurrence in adolescence is the “Alice in Wonderland” phenomenon in which objects are visualized in a very distorted fashion, with exaggerations in size, being either too large or too small. A transverse myelopathy characterized by a sudden onset of profound weakness of the lower extremities and urinary retention may complicate the clinical course of infectious mononucleosis.
Cardiac complications are rare and occur in 1% to 6% of reported series. They usually consist of only nonspecific T-wave changes or minor conduction abnormalities. Myocarditis and pericarditis are rare. Liver function abnormalities are rarely severe, although enzyme changes are common. Primary EBV infection has been associated with a Reye syndrome–like illness. Severe liver dysfunction causing death is a common complication of EBV infection in the X-linked lymphoproliferative syndrome. Hepatic dysfunction is uniformly present with this disorder at the time of death and is the cause of death in about a third of such patients. An unusual manifestation of infectious mononucleosis is intense jaundice. It generally results from a combination of hemolysis and mild hepatitis. Spontaneous rupture of the spleen may occur.
Respiratory difficulties usually consist of upper airway obstruction. Transient interstitial infiltrations, some with effusions, have been recorded. Infectious mononucleosis should be considered in the differential diagnosis of any child with pleural effusions. Renal complications of infectious mononucleosis generally consist of hematuria associated with a mild nephritis. Not all occurrences have been clearly separated from poststreptococcal glomerulonephritis. Severe rhabdomyolysis can be associated on rare occasion with EBV infection. Eye findings in infectious mononucleosis are unusual but may be significant when they do occur. Severe retinochoroiditis is such an ocular complication. In some patients with infectious mononucleosis, lethargy, particularly daytime lethargy, is seen for prolonged periods, often longer than a year.
X-linked lymphoproliferative syndrome is associated with fatal or severe infectious mononucleosis, acquired hypogammaglobulinemia, and malignant lymphoma. Details about X-linked lymphoproliferative syndrome can be found in Chapter 24 .
A variety of malignancies have been linked to EBV, including clonal T-cell proliferations. In patients who undergo transplantation, a spectrum of lymphoproliferative diseases may occur as a result of activation of EBV. These diseases may vary from an infectious mononucleosis–like polyclonal B-cell proliferation to a monoclonal B-cell lymphoma. EBV infection can also result in a hemophagocytic syndrome. The total spectrum of the rare complications and unusual syndromes associated with EBV infection have been reviewed by Timár and colleagues.
Laboratory Diagnosis.
Atypical lymphocytosis is the hallmark of infectious mononucleosis ( Fig. 37-10 ). Several attempts have been made to classify these abnormal cells on morphologic grounds, the best known method being that of Downey and McKinlay. However, it is clear that atypical lymphocytes cannot be easily classified into separate categories and that a spectrum of cell types exists. In general, the atypical lymphocytes of infectious mononucleosis are large, but they vary in size considerably. Their outlines are irregular, and many cells show a characteristic tendency to flow around adjacent erythrocytes. Nuclei are large and usually eccentrically located and pleomorphic, with abundant coarse chromatin and occasional nucleoli. The cytoplasm is generally abundant and typically basophilic. Cytoplasmic vacuoles may be seen. These types of cells are not morphologically specific for infectious mononucleosis and can be seen in other viral infections and after administration of a number of medications.
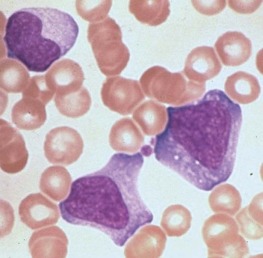
The vast majority of atypical lymphocytes from patients with infectious mononucleosis are thymus derived. These atypical cells possess human T-lymphocyte–specific antigens, as well as sheep erythrocyte receptors. T lymphocytes appear to lack receptors for EBV, and it would appear that only B lymphocytes are infected by this virus. A possible unifying interpretation of the role of T lymphocytes is that these cells represent an immune reaction that protects against this potentially oncogenic virus. Increased numbers of B cells are found during the first week of illness and decline to normal levels in 3 weeks. T lymphocytes reach their peak later, usually 10 to 14 days after the onset of symptoms, and remain elevated for 5 weeks. There may be an early reversal of the ratio of T to B lymphocytes, with a subsequent increase in the percentage of T cells during the second through fifth weeks of illness. It is possible that both T and B cells may be “transformed” into atypical lymphocytes—B cells by infection with EBV and T cells by an immunologic response to viral antigen itself—or the B cells may respond to altered antigens on their surface. EBV-infected B lymphocytes account for only a minority of the atypical lymphocytes found in peripheral blood. In the very early stages of symptomatic illness, however, nearly 20% of all B cells in the circulation may be infected with the virus. The majority of atypical lymphocytes are T lymphocytes. Natural killer (NK) cell activity has been shown to be present during the acute phase of infectious mononucleosis. Interferons, which are inducers of NK cell activity, may have an inhibitory effect on the outgrowth of EBV-infected B lymphocytes in vitro. Significant anergy and diminished lymphocyte responsiveness in vitro to mitogens and antigens exist during the first week of illness. These lymphocyte changes are reflected in a great increase in uric acid turnover, with 55% of infected patients having serum uric acid levels of 8 mg/dL or higher.
Patients with infectious mononucleosis have lymph node pathologic findings that are easily confused with lymphoma. The nodal architecture is distorted by large, dark lymphoid cells, and the capsule may be infiltrated. Reed-Sternberg cells have been reported on several occasions. Pathologic changes are not confined to lymphoid tissue, however. Perivascular cuffing of the brain vasculature, inflammation of the liver, and inflammatory infiltration of the kidney and bone marrow have been repeatedly observed.
The heterophil antibody is so named because the antigen to which the antibody reacts is found in more than one species. The antibody, like anti-i, is an IgM macroglobulin. It agglutinates sheep red blood cells and can be removed completely from serum by preincubation with beef red blood cells but not with guinea pig kidney. Heterophil antibody titers usually increase after the third day of illness, peak at 2 weeks, and may remain positive for several months before ultimately becoming negative (unlike the antibody specific for EBV). This traditional Paul-Bunnell serologic agglutination method has been largely replaced for screening purposes by the spot test, in which finely ground guinea pig kidney or beef red blood cell stroma is added to serum on a slide, followed by a drop of horse cells. Results of the test are considered positive if agglutination occurs in the presence of guinea pig kidney (which absorbs out Forssman antibody but not heterophil antibody) but are negative with beef red blood cell stroma. The spot test requires only 2 minutes and is 96% to 99% accurate. Results of both the Paul-Bunnell test and the spot test are usually negative in preschool-aged children, in whom heterophil antibody production is limited. This age group does produce diagnostic levels of EBV-specific antibody.
EBV is a herpeslike DNA virus. It is a relatively complex virus, and a variety of virus-associated antigens have been described. Antibodies to viral capsid antigen (VCA) and early antigen (EA) are detected early after the onset of EBV-associated infectious mononucleosis. Levels of antibodies to VCA reach their peak at about 3 weeks after the onset of clinical illness. Their levels decline somewhat thereafter, but they remain for life. Antibodies to EA usually last 2 to 4 months. EA has two components that are differentiated by their immunofluorescent staining: “D” for diffuse and “R” for restricted staining. A technique for determining EBV-specific IgM has been described. EBV-specific IgM almost always occurs in the acute phase of infectious mononucleosis. It rarely persists more than 2 to 3 months. During this period, virus shedding from the oropharynx is easily demonstrable.
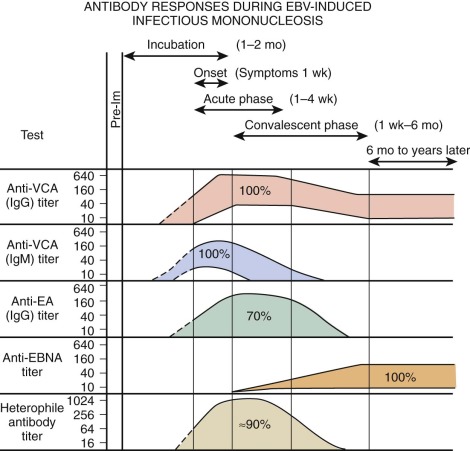
Figure 37-11 shows the characteristic antibody patterns observed in young adults with EBV-induced infectious mononucleosis. Before infection, no antibodies are present. During the acute phase of illness, high titers of IgM and IgG antibodies to VCA are seen. IgM antibodies are transient and disappear after 1 to 2 months. Antibodies appearing against EA disappear after a few weeks to months. Antibodies against Epstein-Barr nuclear antigen are the last to appear and are seen 1 to 2 months after the illness. In young infants, VCA IgM is found in only 60% of patients and EA antibody is identified in only 50%. The only persistent antibody response should be VCA IgG and antibodies to Epstein-Barr nuclear antigen.
Treatment.
There is no evidence that bed rest or rest in general shortens the clinical course of infectious mononucleosis. Patients will determine their own level of activity. The most significant risk during acute illness is splenic rupture, but its incidence is extremely low.
Corticosteroid therapy is often used for infectious mononucleosis. Although these agents may produce improvement of symptoms, enhancement of general well-being, and reduction of fever, their use for these purposes should be restricted. Less controversial is the use of corticosteroid therapy for patients with airway obstruction secondary to tonsillar hypertrophy, severe hemolytic anemia, or thrombocytopenia and bleeding.
Human Immunodeficiency Virus
Epidemiology.
In the early 1980s, a newly described constellation of symptoms that resulted from immune compromise and led to death from opportunistic infections and unusual malignancies was recognized. These early descriptions of what is now known as acquired immunodeficiency syndrome (AIDS) heralded the beginning of what has become one of the major medical, public health, and social issues of our time. Identification of the human immunodeficiency virus (HIV), a retrovirus, as the etiologic agent that transmits the disease has led to major advances in understanding the pathogenesis of AIDS. At least two types of HIV, HIV-1 and HIV-2, exist; both are considered lentiviruses based on similarities in genetic composition, mechanisms of replication, and interaction with their hosts. A characteristic of lentivirus infections is that they cause slowly progressive disease with an incubation period of months to years before clinical symptoms appear. The worldwide spread of AIDS has led to a plethora of information about the biology of the virus and its epidemiology and incidence projections in different countries, as well as a concerted search for effective means of prevention and treatment.
The first occurrences of AIDS in the pediatric population in the United States took place in children born to mothers infected with HIV or in children, especially neonates, who had received blood products contaminated with HIV. The AIDS pandemic in Africa and, in particular, southern Africa has reached catastrophic proportions. The incidence of the disease in eastern Europe, India, and Thailand and throughout most of the Third World poses many political, social, and medical challenges, a discussion of which that is beyond the scope of this chapter.
Transmitted primarily through contact with infected lymphocytes and monocytes, HIV may be found in blood, semen, and vaginal secretions. Isolation of the virus from 13- and 15-week abortuses, placenta, and cord blood provides strong support for intrauterine infection. Maternal-fetal transfusion at delivery is another possible route of infection. Postpartum transmission via breastmilk, although possible, is rare. Accordingly, in countries in which safe feeding substitutes are not readily available, breastfeeding should not be curtailed; however, in developed countries where alternatives exist, breastfeeding by HIV-infected women is discouraged.
The rate of transmission from an untreated mother to child is estimated to be about 25% based on several studies from the United States and Europe. Transmission from mother to child can be significantly reduced by the use of antiretroviral therapy during pregnancy, labor, and the neonatal period.
Major advances in screening and testing of the blood supply in the United States after 1985 led to a reduction in the risk of receiving a contaminated single transfusion in 1988 to 1 in 250,000. The first screening test is a questionnaire to exclude people who engage in high-risk activities. The next step is an enzyme-linked immunosorbent assay (ELISA) for detection of antibodies to HIV antigens. In rare individuals who are infected but seronegative, HIV can escape detection, but sensitive screening tests detect the viral genome by PCR assay. Tragically, individuals with bleeding disorders such as hemophilia A and B who received clotting factors from a large donor pool for a single infusion had an increased likelihood of acquiring AIDS before modern blood screening practice and factor preparation.
Clinical Manifestations.
Infection with HIV results in a spectrum of disease from an asymptomatic state to severe immunodeficiency involving multiple organ systems. The heterogeneity of symptoms is explained by the underlying pathophysiologic course in that (1) the incubation period may be weeks, months, or years; (2) the direct cytopathic effect on target cells, predominantly CD4+ lymphocytes, monocytes, and accessory cells, results in dysregulation of the immune system; and (3) the viral elimination phase that is accompanied by a host response to infection varies with the immune competence of the host. Because many children are infected congenitally, they show signs early in life and often have constitutional symptoms such as unexplained diarrhea, fever, night sweats, generalized lymphadenopathy, and hepatosplenomegaly. Failure to thrive and developmental delay are often prominent early manifestations of HIV infection in children. Susceptibility to recurrent infections by common bacterial and viral pathogens, as well as to opportunistic infections, increases the index of suspicion that the underlying disorder is AIDS. These and other manifestations of HIV infection are summarized in Box 37-1 .
Primary Manifestations
Hematologic and Immune Abnormalities
Hypergammaglobulinemia
Lymphopenia (CD4+ cells decrease)
Decreased CD4/CD8 ratio
Thrombocytopenia
Anemia
Neutropenia
Drug allergies (e.g., trimethoprim in 40% to 60% of patients)
Nonspecific Findings (includes children with two or more unexplained findings for more than 2 months)
Failure to thrive
Hepatosplenomegaly
Generalized lymphadenopathy
Parotitis
Diarrhea (three or more loose stools per day)
Neurologic Disease
Loss of intellectual ability or developmental milestones
Impaired brain growth (acquired microcephaly, brain atrophy, or both)
Progressive systemic motor defects
Paresis
Abnormal tone
Pathologic reflexes
Ataxia or gait disturbance
Cardiovascular Disease
Cardiomyopathy
Arrhythmias
Other Diseases
Hepatitis
Nephropathy (sclerosing glomerulonephritis)
Dermatologic diseases (most commonly seborrheic dermatitis)
Secondary Manifestations
Secondary Infections
Pneumocystis carinii (now Pneumocystis jirovecii ) pneumonia
Chronic cryptosporidiosis
Disseminated toxoplasmosis (onset after 1 month of age)
Extraintestinal strongyloidiasis
Chronic isosporiasis
Candidiasis (esophageal, bronchial, and pulmonary)
Extrapulmonary cryptococcosis
Disseminated histoplasmosis
Mycobacterial infection
Cytomegalovirus infection (onset after 1 month of age)
Coccidioidomycosis
Nocardiosis
Progressive multifocal leukoencephalopathy
Lymphocytic interstitial pneumonitis
Secondary Cancers
Kaposi sarcoma
B-cell non-Hodgkin lymphoma
Primary lymphoma of the brain
The hematopoietic dysfunction that is invariably associated with HIV infection results from global dysregulation of the physiologic cascades of antibody formation, coagulation and complement, and direct infection of key regulatory cells. Acute and chronic infections and therapies for the underlying disorder also affect hematopoiesis. Although for the purposes of discussion it is simpler to view these changes according to cell lineage, it should be stressed that these events rarely occur in isolation. Examination of the peripheral blood smear from the majority of AIDS patients reveals anemia and granulocytopenia and, in a third of patients, associated thrombocytopenia.
Bone Marrow Findings.
In several studies, the bone marrow morphologic changes in patients with AIDS were reported. The most common findings were hypercellularity, lymphoid aggregates, plasmacytosis, and dysplasia. When thrombocytopenia is present, the marrow often contains adequate megakaryocytes, thus suggesting an immune mechanism of platelet destruction. The anemia and granulocytopenia appear to be related to the dysplastic bone marrow and result from ineffective hematopoiesis. Increases in reticulum and fibrosis have been reported in association with Mycobacterium avium infection.
Thrombocytopenia.
The underlying mechanism of thrombocytopenia, which can be the initial finding of HIV infection in children, appears to be immune destruction of platelets. Several studies have identified a high incidence of cytophilic antibodies and have highlighted concomitant complement deposition on the platelet. In addition, a 25-kD platelet-associated antigen has been detected in thrombocytopenic adults and children with AIDS ; however, the presence of the antibody in serum does not invariably lead to thrombocytopenia. Spontaneous recovery from thrombocytopenia does occur; for instance, in one study, 8 of 25 patients with thrombocytopenia recovered.
Adequate treatment of HIV infection with highly active antiretroviral therapy is the first-line approach for HIV-associated ITP and typically improves the thrombocytopenia. ITP-specific treatment is considered when the platelet count decreases to less than 30,000/mL or a clinical bleeding episode is encountered. After HIV-directed treatment, first-line treatment of HIV-associated ITP includes the standard options of prednisone, intravenous immune globulin, and anti-D globulin. Second-line ITP-directed treatment options can be considered with failure of the first-line agents and include splenectomy and thrombopoietin-receptor agonists.
Anemia and Granulocytopenia.
Anemia and granulocytopenia are found in most patients with AIDS and reflect, in large part, ineffective hematopoiesis. The anemia is typically normochromic and normocytic, with a low reticulocyte count and mild to severe anisocytosis and poikilocytosis. Up to 40% of patients with AIDS may have positive findings on a direct Coombs test as a result of absorbed immunoglobulin. Although antibodies have been detected on the surface of granulocytes, the mechanism for granulocytopenia appears to be suppression of bone marrow progenitors. Bone marrow from patients with HIV infection exhibits colony formation (GM-CSF and burst-forming units–erythrocyte) similar to that in HIV-seronegative control subjects when cultivated in serum from an HIV-seronegative donor. However, there is selective suppression of colony formation in bone marrow derived from a patient with HIV infection versus bone marrow from seronegative donors when cultivated in seropositive sera. This suggests that progenitor cells and progeny are able to be infected with HIV and express HIV antigens on their surface that are recognized by HIV antibodies, thus accounting for the suppression. Further support for this hypothesis comes from the demonstration of provirus in megakaryocytes from the bone marrow of patients with AIDS.
Alloantibodies are detected in 30% to 60% of patients with AIDS in different series, with anti-i and anti-I being the most common. Anti-i has also been associated with EBV and cytomegalovirus infections, both of which often coexist with HIV infection and may account for the presence of specific red blood cell antibodies. Antibodies to Le, PL, E, K, Lu, and Sd all have been found, although their relationship to the pathophysiologic course of the anemia in HIV, which in large part does not appear to be hemolytic, remains an open question.
Antiretroviral agents often causes anemia and neutropenia. Macrocytosis with 25- to 40-unit increases in mean corpuscular volume is observed in 74% of patients receiving zidovudine. The macrocytosis is not usually accompanied by anemia, but when it does occur the anemia is usually mild and dose related. In a subgroup of patients, anemia caused by bone marrow suppression is observed. Neutropenia (absolute neutrophil count of 750 to 1000 cells/µL) is seen early in antiretroviral treatment and can be dose limiting.
Laboratory Diagnosis.
Detection of antibodies to HIV antigens is a standard approach to the diagnosis of HIV infection. The first screen is an ELISA; positive results may be confirmed by a specific immunoblot. However, the presence of anti-HIV immunoglobulin of the IgG subclass in an infant does not necessarily indicate that the infant is infected with HIV because maternal IgG crosses the placenta and has a half-life of 28 days. In fact, persistence of maternal IgG has been detected beyond 15 months of age. In contrast, several seronegative HIV-infected children have been reported. Although IgM or IgA anti-HIV antibodies are better indicators of active infection in children because these subclasses of antibody do not cross the placenta, the sensitivity and specificity of the ELISAs for these antibody isotypes are much lower than those for IgG anti-HIV. Definitive proof of HIV infection rests with viral culture and demonstration of the provirus via the very sensitive PCR assay.
Treatment.
Current U.S. treatment guidelines for pediatric HIV infection advocate aggressive therapy with combination antiretroviral regimens. The goal is durable suppression of viral replication with preservation of immune function. The expense and difficulty of adhering to this regimen limit access to the vast majority of infected children in the world. Prevention and education remain the most effective short-term modes of limiting the spread of AIDS.
Infections in the Developing World
A detailed review of the prevalent infections in developing countries, including malaria, visceral leishmaniasis (kala-azar), schistosomiasis, trypanosomiasis, hookworm, bartonellosis, and dengue can be found in Chapter 38 .
Cardiac Disease
The focus of this section is on the three major hematologic complications of cardiac disease: hemolytic anemia, coagulopathy, and increased platelet turnover.
Hemolysis
A number of instances of continuing hemolysis and progressive anemia have been reported after the insertion of prosthetic valves, particularly in the aortic area. These conditions may also occur postoperatively when intracardiac patches have been placed (the “Waring blender” syndrome). They are also being recognized as an increasing problem after endoluminal closure of the ductus arteriosus and occasionally after repair of a ventriculoseptal defect. The mechanism of such erythrocyte destruction has been related to failure of endothelialization of patches, thrombosis or perforation of prosthetic valves, and improper placement of prosthetic valves, especially when insufficiency develops at the suture lines. Erythrocyte destruction and ensuing hemolysis, however, have been described in the absence of these complications and been attributed to red blood cell mechanical trauma associated with apparently normal function of the prosthetic valve.
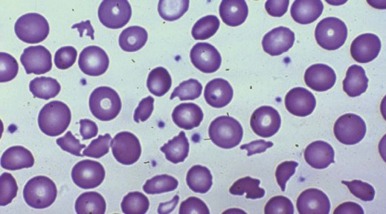
Red blood cell survival studies have clearly shown that prosthetic valve and patch hemolysis is due to an extracorpuscular defect. In general, hemolytic anemia results from fragmentation of the red blood cells as they are mechanically “battered” against a distorted vascular surface ( Fig. 37-12 ). In some instances, it has been postulated that the red blood cell fragmentation may be caused by contact with fibrin deposited in small blood vessels as a result of localized intravascular coagulation. Most times, however, hemolysis is the result of direct mechanical trauma. Nevaril and associates demonstrated that a shearing stress of 300 dynes/cm 2 causes hemolysis in vitro whereas less stress may result in deformed red blood cells morphologically similar in appearance to cells in cardiac hemolytic anemia and microangiopathic hemolytic anemia.
The consequence of the process is hemolytic anemia of the intravascular type associated with hemoglobinemia and hemoglobinuria. Iron deficiency develops quickly in patients with this disorder as a result of increased loss of body iron in the form of hemosiderin, which is shed within the renal tubular cells into urine. The onset of iron deficiency may be of clinical importance. The microcytic hypochromic cell in iron deficiency is more rigid, thereby leading to an accelerated rate of hemolysis from mechanical shearing in the microvasculature. Plasma haptoglobin levels fall. Large quantities of red blood cell lactate dehydrogenase are released into serum, and a close correlation exists between the logarithm of serum lactate dehydrogenase and the half-life of chromium-labeled erythrocytes. The rate of hemolysis may or may not result in anemia. If surgical correction of the defect causing hemolysis is not possible, the patient should be treated with both iron and folate, as well as red blood cell transfusion. The latter is intended to correct anemia, reduce stroke volume, and presumably reduce shear force. Sears and Crosby observed that the severity of hemolysis is directly related to physical activity. This finding has been used as a test to determine whether the hemolysis is of cardiac origin because, if it is, rest also diminishes the rate of hemolysis. If the source of the problem cannot be corrected surgically, propranolol, which reduces the shearing stress between red blood cells and the vascular wall by slowing the velocity of the circulation, may be given.
Mechanical injury to red blood cells may result in loss of pieces of cell membrane, with or without loss of hemoglobin. Such loss will lead to the formation of spherocytes. Consequently, in many patients with heart valve hemolysis, results of the red blood cell osmotic fragility test may be abnormal. Mechanical destruction of red blood cells on abnormal surfaces of the vasculature can result in hyperkalemia. This has been reported to cause ventricular arrhythmias.
Occasionally, AIHA is observed after cardiac surgery involving the placement of foreign material within the vascular system. Children with 22q11.2 deletion syndrome, often with underlying congenital heart disease, are at higher risk for AIHA.
Erythrocyte disturbances in children with cardiac disease must be understood in the context of compensatory states or the context of iron deficiency as a complication. For example, infants with cyanotic congenital heart disease have erythropoietin-induced compensatory polycythemia. Aortic oxygen saturation higher than 80% is usually associated with low erythropoietin titers and hemoglobin levels that will not cause hyperviscosity. Even with moderate degrees of hypoxemia, elevated erythropoietin levels are not seen; presumably, the modest elevation in hemoglobin levels provides adequate tissue oxygenation. Infants with cyanotic congenital heart disease have higher iron requirements because of the greater hemoglobin mass. Diminished iron stores in patients with cyanotic congenital heart disease are associated with a more right-shifted oxyhemoglobin dissociation curve. Most children with cyanotic congenital heart disease have evidence of mild macrocytosis. A mean corpuscular volume greater than the 90th percentile for age and sex nearly eliminates the possibility of iron deficiency.
Acquired Coagulation Abnormalities
Many investigations have suggested that a coagulopathy exists in some patients with cyanotic congenital heart disease. Thrombocytopenia, low plasma fibrinogen levels, defective clot retraction, hypoprothrombinemia, factor V and VIII deficiency, and evidence of fibrin degradation products in serum have been reported. The presence of coagulation abnormalities may correlate with the extent of polycythemia. Marked derangements in coagulation often accompany surgery involving cardiopulmonary bypass. Conflicting data suggest that the coagulation defects associated with cyanotic congenital heart disease must be multifactorial in origin and may be associated with both a bleeding and prothrombotic predisposition. Therefore, each child should be studied individually.
Acquired von Willebrand disease is well recognized in children with congenital heart disease as well as children with aortic stenosis, with ventricular-assist devices, and on extracorporeal life support. Mechanical destruction of high-molecular-weight multimers occurs owing to increased shear force within the abnormal cardiac anatomy. Acquired von Willebrand disease causes a mucocutaneous and/or gastrointestinal bleeding predisposition. Laboratory results will demonstrate decreased von Willebrand factor collagen binding and decreased large von Willebrand factor multimers. Treatment includes supportive care with von Willebrand factor/factor VIII concentrate and/or desmopressin as a bridge to correcting the underlying contributing cardiac defect.
Repair of congenital heart disease can also be associated with coagulopathy. The Fontan operation is the final surgery in the staged palliation of different forms of congenital heart disease with a univentricular heart. After the Fontan procedure, there is often increased systemic venous pressure that causes hepatic dysfunction and/or protein-losing enteropathy, both of which are associated with abnormalities of coagulation.
Management of the hemostatic defects is not settled. It is agreed that they may predispose patients to postoperative hemorrhage, but they are rarely associated with preoperative clinical bleeding tendencies. Suggested management procedures have included the use of heparin to lower viscosity, erythrocytapheresis with plasma exchange, and aminocaproic acid to inhibit fibrinolysis. Whether any of these modalities are indicated and, indeed, whether they might be uniformly effective is speculative. If erythrocytapheresis is chosen, the procedure should be done with great care in cyanotic patients. Withdrawal of red blood cells must be accompanied by infusion of an equal volume of fresh-frozen plasma. Simple removal of red blood cells without volume replacement in polycythemic individuals may cause an acute increase in viscosity, vascular collapse, seizures, and even stroke.
Because of the high incidence of cerebrovascular accidents in children with cyanotic congenital heart disease, some have believed that a hypercoagulable state exists. Hyperviscosity alone may account for some ischemic infarctions. The clinician should be alert to the possibility of iron deficiency predisposing patients to stroke in the presence of polycythemia. Several children in whom neurologic deficits developed after the onset of iron deficiency in the presence of polycythemia have been described. Card and Weintraub showed that red blood cells from animals with iron deficiency have decreased deformability. Altered deformability in the presence of increased blood viscosity could theoretically result in vascular ischemia.
Platelet Abnormalities
Quantitative and qualitative platelet abnormalities are commonly associated with cardiac disease. Certain syndromes, such as DiGeorge syndrome, Noonan syndrome, and Jacobsen syndrome, are associated with both congenital heart disease and platelet abnormalities. However, even in those without a diagnosed genetic syndrome, platelet abnormalities are commonly found.
In one series, the mean platelet count in cyanotic patients with an arterial oxygen saturation of less than 60% was 185,000 cells/mm 3 , as compared with 315,000 cells/mm 3 in patients with an arterial oxygen saturation greater than 60%. These abnormalities are readily noted on preoperative evaluation of the coagulation status of individuals about to undergo surgery. Examination of bone marrow has failed to demonstrate any quantitative changes in megakaryocytes to account for these platelet differences. This, together with the finding of shortened platelet survival in many patients, has suggested that the mechanism of the thrombocytopenia is destructive. Shortening of platelet survival with or without thrombocytopenia has been observed in both children and adults with prosthetic heart valves. The increased platelet turnover may result from a combination of mechanical damage to the platelet and adhesion to the foreign material. Some patients, especially those with minimal cyanosis, may have elevated platelet counts. Although iron deficiency is commonly associated with cyanotic congenital heart disease, the quantitative platelet abnormalities are not related to iron status.
Qualitative platelet defects associated with cyanotic congenital heart disease may include abnormal aggregation in response to adenosine diphosphate, epinephrine, and collagen. In as many as 70% of patients, either a delayed or no second wave of platelet aggregation is observed. These platelet functional abnormalities appear to be due to a defective platelet release mechanism because diminished release of carbon-14–labeled serotonin occurs in response to adenosine diphosphate whereas uptake of it is normal. Platelet release abnormalities are more common in patients older than 4 years, in those with a hematocrit greater than 60%, and in those with platelet counts less than 175,000 cells/mm 3 .
Miscellaneous Hematologic Manifestations of Heart Disease
Subacute bacterial endocarditis may be associated with a variety of hematologic manifestations. Anemia is often present and is usually the result of chronic infection. The white blood cell count is generally normal, although marked leukocytosis or leukopenia may occur. A 25-year review of the evolving pattern of pediatric endocarditis shows that anemia is present in 40% of children with endocarditis and leukocytosis in 30% of such patients. Pancytopenia has been reported, and thrombocytopenia has been noted in a few patients.
Congestive heart failure may result in sufficient hypoxia to cause nucleated red blood cells to appear in the peripheral blood in association with mild reticulocytosis. Thrombocytopenia may be present but is almost exclusively the result of hypersplenism.
Cyanotic heart disease may result in poor perfusion of the spleen and subsequent functional hyposplenism manifested by Howell-Jolly bodies in peripheral blood. This finding is likely to lead to the mistaken conclusion that a child may have the asplenia syndrome (absence of the spleen, cardiovascular malformations, abdominal situs inversus, and other anatomic malformations), which is a separate and distinct diagnosis.
Pulmonary Disease
Hypoxia may be caused by a wide variety of pulmonary disorders and, in turn, results in a form of secondary polycythemia and compensatory shifts in the hemoglobin-oxygen dissociation curve that can be attributed to an increase in the red blood cell content of 2,3-diphosphoglycerate (2,3-DPG). Rarely, the polycythemia results in a state of extreme hyperviscosity and decreased tissue blood flow. Other than polycythemia, the hematologic findings of specific pulmonary disorders tend to be unique to those disorders.
Cystic Fibrosis
Patients with cystic fibrosis demonstrate a significant impairment in erythropoietic response to hypoxemia. There is neither an appropriate increase in hemoglobin level nor an adequate shift in the red blood cell–oxygen affinity curve. Failure of the hemoglobin-oxygen dissociation curve to compensate means that such patients may be more symptomatic at any given hemoglobin level than patients who have anemia and other disorders. Disturbances in erythropoietin regulation, as seen in the anemia of chronic disorders and iron deficiency, appear to be the principal cause of the relative anemia in children with cystic fibrosis. Deficiencies in vitamin E and iron are also common. If a severe hemolytic anemia is observed in a subject with cystic fibrosis, it may be related to vitamin E deficiency.
Idiopathic Pulmonary Hemosiderosis
Idiopathic pulmonary hemosiderosis (IPH) is an uncommon chronic disease that usually affects children and young adults. However, the age at onset may be as early as the neonatal period. IPH is characterized by recurrent intrapulmonary hemorrhage and may result in hemoptysis and pulmonary insufficiency. The hematologic manifestation of this disorder is iron-deficiency anemia.
The etiology of IPH is not known. A hereditary or familial tendency has been suggested. An immunologic cause seems likely because other occasional findings include positive Coombs test results, the presence of cold agglutinins, and an increased number of mast and plasma cells in the lungs. Autoimmunity is also suggested by frequent association with celiac disease, vasculitis, and collagen vascular disease, as well as by response to treatment with immune suppression. Historically, an association with hypersensitivity to milk has been suggested as well as a structural defect in the alveolar capillaries.
The most helpful clinical signs for identifying IPH are iron-deficiency anemia and recurrent or chronic cough, hemoptysis, dyspnea, wheezing, and, frequently, cyanosis. Any single feature may be present without the others. For example, occasionally the only clinical sign is iron-deficiency anemia. Pulmonary symptoms may be present without radiologic findings and vice versa. When pulmonary symptoms are predominant, there may be associated fever, tachycardia, tachypnea, leukocytosis, an elevated sedimentation rate, and, occasionally, abdominal pain. Radiographic abnormalities vary from minimal transient infiltrates to massive parenchymal involvement with atelectasis, emphysema, and hilar adenopathy.
The anemia reflects an iron-deficiency state resulting from excessive accumulation of iron in the lungs. This iron is usually sequestered in alveolar macrophages and is largely unavailable for new red blood cell formation. With time, however, the iron is eventually lost from the lungs. In some patients, iron administration fails to correct the anemia. This is usually a reflection of inadequate heme synthesis resulting from the anemia of chronic disease. AIHA may occur. Eosinophilia is found in 15% to 20% of children with IPH.
A diagnosis of IPH may be made by the finding of siderophages in the gastric aspirate. Siderophages will stain positive with Prussian blue dye. Sputum evaluation and bronchoalveolar lavage may reveal hemosiderin-laden alveolar macrophages. If the diagnosis of IPH is seriously being considered, it may be necessary to perform a lung biopsy. Typical findings include alveolar epithelial hyperplasia, degeneration with excessive shedding of cells, large numbers of siderocytes, varying amounts of interstitial fibrosis and mast cell accumulation, elastic fiber degeneration, and sclerotic vascular changes. Electron microscopic examination shows no evidence of subendothelial deposits or basement membrane lesions. No evidence for localization of IgG, IgM, C1q, or fibrinogen has been found. The results of needle aspiration or needle biopsy may also provide a diagnosis.
Treatment of IPH continues to be a controversial subject due to its rarity. In those patients with concomitant celiac disease, a gluten-free diet is recommended. Given the historic link with hypersensitivity to cow’s milk, a trial of removal of cow’s milk from the diet is appropriate in some instances. Corticosteroid therapy will sometimes produce a remission of disease activity. Immunosuppressive therapy may be useful, but the results of such treatment are unpredictable.
Pulmonary Hemosiderosis from Other Causes
Clinical findings of hypochromic and microcytic anemia and pulmonary infiltrates may be seen in association with glomerulonephritis (Goodpasture syndrome), collagen vascular disease, Wegener granulomatosis, and, occasionally, SLE. Episodes of pulmonary hemosiderosis have also been described in patients with cystic fibrosis and celiac disease.
Hematologic Findings in Other Pulmonary Disorders
Sarcoidosis does not usually cause specific hematologic disturbances, although anemia is present in some patients. Sarcoidosis can be associated with splenic enlargement in up to 25% of patients, often with signs of hypersplenism with anemia, leukopenia, and thrombocytopenia. A higher than average incidence of blood group antigen A has been reported in this disorder. Bone marrow granulomas are noted occasionally. The significance of this finding is not known, but the altered immune response of these patients and an association of sarcoidosis with hemolytic anemia and immune thrombocytopenia suggest that sarcoidosis represents a generalized autoimmune disorder.
Eosinophilia may be observed in a variety of pulmonary disorders, including asthma, Löffler syndrome, tropical pulmonary eosinophilia, polyarteritis nodosa, and sarcoidosis.
Renal Disease
Renal disease may produce disturbances in red blood cells, white blood cells, platelets, and coagulation factors. In many instances, the abnormalities that are found do not parallel the status of renal function but rather reflect the activity of the disease process that results in renal dysfunction. The hematologic aspects of renal insufficiency have been reviewed in detail.
The Red Blood Cell
Anemia
Anemia is a common finding in acute and chronic renal disease and is often so severe in these conditions that it constitutes the major morbidity of renal disease. Although the anemia is often multifactorial, the main cause is erythropoietin deficiency. Progressive renal disease is accompanied by an erythropoietin decline that results in a hypoproliferative, usually normochromic, normocytic anemia. However, other factors can intervene to impair erythropoietin responsiveness and aggravate already low levels of the hormone. These include infection and inflammation (anemia of chronic disease), inadequate dialysis, and secondary hyperparathyroidism. In patients treated with erythropoietin, deficiencies of cobalamin (vitamin B 12 ), folic acid and, especially, iron may limit response. Transfusion support frequently becomes a necessary intervention, although elevated 2,3-DPG levels and a right-shifted oxygen dissociation curve make the red blood cells of uremic subjects more effective in the delivery of oxygen than normal red blood cells. The anemia usually develops when the creatinine clearance decreases below 35 to 45 mL/min, although no direct correlation is found between the severity of the anemia and the degree of renal impairment. The hemoglobin level is generally in the 7- to 8-g/dL range in uremic states.
Decreased red blood cell production is the major cause of the anemia of chronic renal failure. Although deficiency in erythropoietin production is the primary cause of decreased red blood cell production in renal failure, suppression of hematopoiesis by serum inhibitors may be a contributing factor. In patients with acute or chronic renal failure as a result of nephrectomy, normal or even increased erythropoietin production can be maintained. Several series have documented improved hematocrits and erythrocyte volumes proportional to the intensity of dialysis treatments, which may remove the inhibitors.
In addition, excess parathyroid hormone is also an inhibitor of erythropoiesis in uremia, a defect that renal failure states share with primary hyperparathyroidism. The mechanism of this contribution to anemia is likely due to the replacement of the marrow cavity by fibrous tissue in hyperparathyroid states. Removal of hypertrophied parathyroid glands improves the osteitis fibrosa of involved marrows and the anemia that accompanies this abnormality. Treatment with 1,25-dihydroxycalciferol also decreases marrow fibrosis and diminishes the anemia of renal failure. The role of parathyroid hormone in these anemias is in remodeling the environment in which hematopoiesis takes place; the more advanced states of bone disease seen in patients with uremia may even lead to splenomegaly secondary to extramedullary hematopoiesis.
In renal disease, unlike many other chronic disease states, close correlation exists between erythrocyte mass and erythrocyte survival. This implies that shortening of red blood cell survival has an important role in the development of anemia in these patients. About 70% of patients with renal disease have shortened red blood cell survival related to a host of potential alterations. These cells demonstrate increased mechanical fragility, autohemolysis, diminished deformability, and a variety of metabolic defects.
A circulating inhibitor of red blood cell metabolism accumulates in some but not all uremic patients. This inhibitor diminishes recycling of glucose through the hexose monophosphate shunt, a defect that may be clinically important. There are several reports of severe hemolytic disease in uremic patients given sulfa drugs. Because of this problem, caution in the use of oxidant drugs is advised in those with azotemia. The decreased shunt activity may be secondary to a metabolic defect within the shunt itself or due to a block within the main glycolytic pathway. Other incidental causes of hemolysis may be observed during hemodialysis. Contaminants such as copper, nitrate, and chloramines in hemodialysis baths may cause varying degrees of hemolysis.
The erythrocytes of patients with renal disease are generally normochromic and normocytic without any distinguishing morphologic characteristics. Occasionally, scalloped or burr cells are observed, usually in association with uremia secondary to specific causes or related to specific syndrome complexes such as hemolytic uremic syndrome ( Fig. 37-13 ). In diseases associated with microangiopathic hemolytic anemia, red blood cell fragmentation is common. It may be observed in patients with malignant hypertension, renal cortical necrosis, polyarteritis nodosa, hemolytic uremic syndrome, SLE, and TTP.
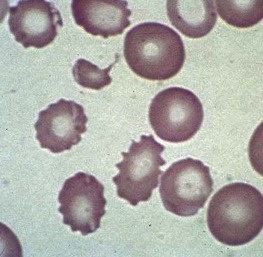
Anemia associated with megaloblastic changes in bone marrow is not uncommon in chronic renal failure. This anemia is usually due to folate deficiency caused by poor dietary intake, hemodialysis, peritoneal dialysis, or the effects of immunosuppressive drugs. In addition, defective protein-mediated folate transport may be present. Serum from uremic patients may contain an increased concentration of folic acid–binding protein. Folate deficiency is usually suspected when large numbers of macroovalocytes are seen in association with hypersegmentation of polymorphonuclear leukocytes on the peripheral blood smear. The anemia of renal disease can also be worsened in dialyzed patients by superimposition of iron deficiency due to blood loss during hemodialysis.
The primary role of erythropoietin deficiency in the anemia of renal failure has been incontrovertibly confirmed with the successful trials of recombinant human erythropoietin in both dialyzed and nondialyzed renal failure patients (see Chapter 1 ). Administration of the hormone has eliminated the need for red blood cell transfusions or androgens, previously the mainstays in treating the anemia of renal failure. The therapeutic success of erythropoietin-stimulating agents (ESAs) has brought to a close any doubts concerning the major cause of the anemia of renal failure. The administration of ESAs may unmask iron and folate deficiency, their presence being signaled by an apparent refractoriness to erythropoietin administration. Controversy exists about the goal hemoglobin level in patients with renal insufficiency treated with ESAs, although most data support a lower hemoglobin level of 10 to 11 g/dL. Administering ESAs to reach higher target hemoglobin levels greater than 13 g/dL is associated with fewer red blood cell transfusions; however, higher hemoglobin levels are also associated with an increased risk for stroke, hypertension, and thrombosis. It remains unclear whether the higher hemoglobin is linked to these adverse outcomes or high doses of ESAs.
Polycythemia
Secondary erythrocytosis is a well-recognized complication of renal disorders. The increase in red blood cell mass is due to release of erythropoietin directly from renal tumor cells, renal ischemia, and hydronephrosis and in association with chronic glomerulonephritis, pyelonephritis, nephrosclerosis, and nephrotic syndrome in adults. Erythrocytosis is also a common finding after renal transplantation.
The White Blood Cell
Azotemia and uremia per se appear to have relatively little effect on white blood cell function. Granulocytopenia is common during hemodialysis and appears to be due to transient sequestration of granulocytes in the pulmonary vascular bed followed by later release back into the circulation. After hemodialysis, a transient augmentation in granulocyte adherence coincides with the time of sequestration of the pulmonary vascular bed. The adherence-augmenting factor in the plasma of patients undergoing dialysis is heat stable.
Neutropenia as a result of folate deficiency may be present in patients with renal failure. The cause of the neutrophil nuclear hypersegmentation associated with uremia is unknown and should not necessarily be attributed to folate deficiency.
Vascular occlusion of the kidney may result in eosinophilia. Approximately 80% of adult subjects with embolic renal disease have eosinophil counts ranging from 540 to 2000 cells/mm 3 .
Coagulation Abnormalities
Patients with uremia may have varied changes in some coagulation factors. Among the most common abnormalities observed are mild to moderate depressions in factors V, VII, IX, and X. These abnormalities are related to the high incidence of hepatic dysfunction in patients with uremia or vitamin K deficiency. The level of fibrinogen is regularly elevated because it is an acute phase reactant. Increased fibrinogen turnover has been demonstrated in experimental models, and excessive fibrin degradation products have been found in the urine and serum of uremic patients. When a decrease in one or more consumable clotting factors (factors I, II, V, or VIII) is also present, it may signify the existence of consumptive coagulopathy. Because fibrin deposition may occur in the absence of such a coagulopathy, it is often helpful to determine whether high-molecular-weight fibrin degradation products are present in the urine and serum. If found only in the urine, such products are probably derived from the dissolution of fibrin deposits in the kidneys. If demonstrable simultaneously in urine and serum, their presence indicates renal injury because such degradation products cannot be demonstrated in the urine of patients with streptokinase-induced fibrinolysis who have no renal disease. Fibrin degradation products in the urine and serum usually indicate active renal disease, although other reasons for this finding may be seen in patients with renal disease. Fibrin degradation products may also result from concomitant liver disease or from an external shunt used for hemodialysis. Urinary fibrinolytic activity is either diminished or absent in renal failure, and inhibitors of urokinase-induced plasminogen activation are demonstrable.
Nephrotic syndrome is often accompanied by changes in a number of coagulation factors. Increased levels of fibrinogen, factor VIII, and the factor VII-X complex are commonly present. A prolonged partial thromboplastin time is often observed and is usually due to a decrease in the plasma level of factor IX. Factor IX is consistently found in urine containing more than 10 g of protein per 24 hours in patients with nephrotic syndrome. Plasma levels of factor IX rarely decrease to less than 10%, and clinical bleeding episodes are unusual. With resolution of the proteinuria in patients receiving corticosteroid therapy, levels of factor IX increase. Factors II and VII have also been identified in considerable quantity in the urine of patients with nephrotic syndrome despite normal levels of these factors in plasma. This finding suggests that loss of some coagulation factors into urine can be fully compensated for by an increased rate of synthesis. A prolonged partial thromboplastin time may also be due to a low level of Hageman factor (factor XII). Hageman factor has a molecular mass of approximately 75,000 kD, similar to that of albumin, and losses in urine are to be expected. Although factor XII levels may decrease to less than 10%, as with congenital deficiency of this factor, bleeding is not encountered. Patients with nephrotic syndrome appear to have a substantially higher risk for thromboembolic complications. The coagulation defects associated with nephrotic syndrome have been reviewed by Kaufmann and colleagues.
The hypercoagulable state seen in many forms of renal disease may be compounded by the occurrence of lupuslike anticoagulants. This is common in a variety of end-stage renal diseases, with a rate of occurrence as high as 22%. Whether therapy focused on prevention of fibrin deposition within the kidney is of benefit to patients with certain forms of glomerulonephritis remains uncertain. Kincaid-Smith used a combination of an anticoagulant, dipyridamole, and cyclophosphamide in a series of patients with membranoproliferative glomerulonephritis. A similar approach was reported by Robson and associates, but with azathioprine substituted for cyclophosphamide. In both studies, it was suggested that these therapies produced improvement in the patients’ clinical course. The concept of anticoagulation therapy for these disorders has been reviewed.
Platelet Abnormalities
Mild thrombocytopenia occurs in approximately 25% of patients with acute renal failure and 10% of those with chronic renal disease. The thrombocytopenia is most likely due to impaired production, because platelet life span in uremia is generally normal.
Platelet function is abnormal in patients with renal failure. The standardized Ivy bleeding time is usually prolonged and generally correlates with the clinical tendency for bleeding in these patients. Platelet adhesion and platelet factor 3 availability are diminished in acute and chronic uremia. Impaired aggregation of platelet-rich plasma in the presence of adenosine diphosphate, epinephrine, thrombin, and collagen has been observed. Clinical bleeding episodes related to these platelet abnormalities can usually be managed with desmopressin given intravenously. Long-term administration of recombinant erythropoietin also improves the biochemical and functional changes in the platelets of uremic patients. Rarely, enhanced platelet function characterized by hyperaggregability has also been observed in uremia.
Platelet functional abnormalities are not due to an intrinsic platelet defect because dialysis quickly reverses these changes and incubation of normal platelets in plasma from uremic patients promptly induces functional aberrations. Rather, platelet dysfunction appears to be due to the interaction of intrinsically normal platelets with various abnormal dialyzable metabolites found in the plasma of uremic patients. If urea and creatinine levels are elevated, some of these findings can be reproduced. Phenols and guanidinosuccinic acid, which accumulate in the plasma of uremic patients, can also account for platelet functional abnormalities. These findings are discussed in detail in Chapter 34 .
Several hereditary disorders have been reported in which quantitative or qualitative platelet abnormalities occur in association with nephritis. MYH9 disorders are caused by defects in the myosin heavy chain IIa and associated with macrothrombocytopenia and a bleeding tendency. Because myosin IIA is a major component of the podocyte foot process and maintains capillary wall integrity, many MYH9 disorders, including Epstein syndrome, Fechtner syndrome, and Sebastian platelet syndrome, are associated with kidney dysfunction in addition to hearing loss and cataracts. In these disorders it is hypothesized that disordered early platelet release contributes to the thrombocytopenia. The inherited platelet disorders are discussed in Chapter 30 .
The pathophysiology and management of hemolytic uremic syndrome are discussed in Chapter 34 .
Gastrointestinal Disease
Hematologic complications of gastrointestinal disease appear in many disorders. This section does not include the wide range of situations in which blood loss from the gastrointestinal tract occurs but rather focuses on specific diseases and their hematologic complications.
Diseases of the Gastrointestinal Tract
Esophagus
Plummer-Vinson syndrome (dysphagia, postcricoid webs, and iron deficiency) occurs in older individuals and is discussed in Chapter 11 . Iron-deficiency anemia may be the only manifestation of gastroesophageal reflux; thus endoscopy is important in the evaluation of unexplained iron-deficiency anemia. Chronic gastroesophageal reflux may cause Barrett esophagus with its attendant risk for the development of adenocarcinoma.
Stomach
The gastric mucosa is important in both vitamin B 12 and iron absorption, and disorders of the gastric mucosa may cause defective absorption of either of these nutrients. The gastrointestinal causes of iron and vitamin B 12 deficiency are reviewed in Chapters 10 and 11 .
Gastric resection may result in iron or vitamin B 12 deficiency, the former from bleeding at sites of anastomosis and the latter, years later, from lack of intrinsic factor. Vitamin B 12 deficiency may develop in infants breastfed by women who have undergone gastric or intestinal bypass procedures. Macrocytic, megaloblastic anemia resulting from vitamin B 12 deficiency has been reported in association with gastric trichobezoars. This is presumed to be due to bacterial overgrowth of the upper gastrointestinal tract. In congenital intrinsic factor deficiency, gastric biopsy results are normal and no antibodies to parietal cells or intrinsic factor are present.
Small Intestine
Celiac disease and inflammatory bowel disease are both frequently associated with anemia and are covered in detail in Chapter 11 .
Crohn disease has also been associated with coagulopathy, particularly with acute flares. Low factor XIII levels have been observed in active disease associated with regional enteritis. An abnormality in arachidonic acid metabolism may occur in patients with chronic inflammatory bowel disease, including regional enteritis. This may result in alterations in platelet function. Diminished neutrophil function may be seen in both regional enteritis and other forms of inflammatory bowel disease. The condition may be identified by decreased oxidative metabolism and low superoxide dismutase content of neutrophils. Perinuclear antineutrophil cytoplasmic antibodies are commonly found in patients with inflammatory bowel disease. Inflammatory bowel disease, particularly during acute flares, is also associated with an increased risk for thrombosis.
Disorders such as tropical sprue, celiac disease, bowel lymphoma, amyloidosis, and connective tissue disturbances (including Ehlers-Danlos syndrome and pseudoxanthoma elasticum) may produce panselective or selective malabsorption. Certain disorders such as intestinal lymphangiectasia may cause protein loss of a magnitude sufficient to impair globin chain synthesis. Protein loss may also be associated with selective loss of T lymphocytes into the bowel lumen, which causes lymphopenia and altered delayed-type hypersensitivity. This lymphopenia may be associated with hypogammaglobulinemia.
Eosinophilic gastroenteritis is a disorder most often characterized by recurrent bouts of abdominal pain, nausea, vomiting, diarrhea, and an elevated peripheral blood eosinophil count for which there is no other explanation. The gut wall may be infiltrated by eosinophils.
Common diarrheal illnesses during infancy may result in life-threatening methemoglobinemia, especially if the diet has been high in nitrates.
Large Intestine
Ulcerative colitis is often associated with iron-deficiency anemia as a result of blood loss. AIHA identified by positive results of a Coombs test and immune thrombocytopenia are relatively rare complications of ulcerative colitis, however, and develop in less than 1% of patients. Occasionally, vitamin B 12 deficiency occurs after many years if a “backwash” ileitis is present. Polymorphonuclear leukocytosis is common. Occasionally, an increase in plasma fibrinolytic activity will be observed. A significant decrease in antithrombin III levels may be found. Some clinicians have treated this complication with ε-aminocaproic acid. Platelet functional abnormalities may exist and are discussed in Chapter 34 . Alterations in T-cell subsets may also occur in children with inflammatory bowel disease.
Constipation has been reported to cause an increase in sulfmethemoglobin levels. It has been suggested that excess nitrite absorption from the gut may be present in some patients as a result of abnormal bowel function.
Symptoms of Peutz-Jeghers syndrome (gastrointestinal polyposis and mucocutaneous pigmentation) may occur in early childhood and are associated with an increased risk for intestinal cancer.
Hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu disease) may result in iron deficiency from gastrointestinal bleeding. Alterations in hemostasis (DIC, platelet dysfunction, and factor XI deficiency) have been reported. An association between this disorder and von Willebrand disease may exist.
Disease of the Pancreas
Acute hemorrhagic pancreatitis may cause acute anemia during the first week of the illness as a result of hemodilution, intravascular coagulation, and blood loss. Leukocytosis with neutrophilia is a usual finding. Markedly elevated levels of fibrin split products usually suggest that a consumptive coagulopathy is present. A concomitant decrease in platelet count is also common. High levels of methemalbumin are common in the ascitic fluid of individuals with hemorrhagic pancreatitis and can be used to distinguish this disorder from nonhemorrhagic pancreatitis. Patients with ascitic fluid are the ones most likely to have evidence of consumptive coagulopathy. For example, fibrin degradation products are seen in 40% of subjects with fulminant pancreatitis without ascites but in 100% of patients with clinically apparent peritoneal fluid.
Shwachman-Diamond syndrome and Pearson syndrome with associated congenital exocrine pancreatic insufficiency and bone marrow failure are discussed in Chapter 7 .
Cystic fibrosis may cause malabsorption, which produces the expected hematologic abnormalities. A specific pattern of anemia in association with edema and hypoproteinemia may be observed in some children with cystic fibrosis. It is found most commonly in children who have low dietary intake of nitrogen, such as those who receive their protein primarily from breastmilk or soybean formula. The edema is caused by hypoalbuminemia. The anemia responds to adequate dietary protein enrichment and pancreatic enzyme supplementation but not to iron administration. Cystic fibrosis may also be associated with malabsorption of fat-soluble vitamins, and the consequent coagulopathy is related to vitamin K deficiency. Vitamin E deficiency may also occur and is manifested by mild hemolytic anemia and abnormal red blood cell peroxide hemolysis, as well as by hyperaggregation of platelets.
Diseases of the Liver
Red Blood Cell Disturbances
Anemia is common in acute and chronic liver disease and has a diverse etiology. Liver disease is often accompanied by increased portal pressures, with the resultant congestive splenomegaly potentially creating any combination of anemia, leukopenia, or thrombocytopenia. The marrow’s compensation for such hemolytic insults is frequently dampened or absent in the setting of liver disease because of concomitant vitamin nutritional deficiencies. In addition, portal hypertension and liver disease are associated with increased risk for bleeding and concomitant iron deficiency. Shortened red blood cell survival may be observed in some patients with no evidence of blood loss. The exact mechanism by which red blood cell survival is shortened is unclear.
Erythrocytes from patients with active liver disease have an increased tendency to form Heinz bodies after incubation with an oxidant chemical such as acetylphenylhydrazine or sodium ascorbate. This increase in Heinz body formation is associated with increased instability of red blood cell reduced glutathione, decreased hexose monophosphate shunt activity, and reduced glucose recycling through the hexose monophosphate shunt. Activation of superoxide dismutase and glutathione reductase may be observed in the red blood cells of patients with profound liver disease. This may partially explain the alterations in the antioxidant system noted with these disorders. The hemolytic anemia associated with liver disease secondary to Wilson disease is especially severe. The same is true of the hemolytic anemia related to protoporphyria.
The exact significance of these metabolic alterations has not yet been determined. Red blood cell metabolism appears to return to normal within a few weeks after an insult to the liver. All of the metabolic consequences of liver disease can be reproduced in normal intact red blood cells by prolonged incubation in plasma from patients with active liver disease. In patients with active liver disease, it may be wise to withhold any drug that has the potential to present an oxidant challenge to the red blood cell. Similar acquired abnormalities in hexose monophosphate shunt activity have been associated with drug-induced hemolysis in patients with uremia.
The red blood cells in liver disease are often macrocytic, with mean corpuscular volumes in the range of 100 to 110 fL. In patients with biliary obstruction, the red blood cell surface lipids increase and cause target cells. With severe hepatocellular damage, acanthocytes or spur cells are commonly observed ( Fig. 37-14 ). These morphologic abnormalities appear to be in direct proportion to the increase in red blood cell membrane cholesterol that accompanies hepatocellular liver disease. Increased osmotic resistance is a consequence of the increased red blood cell surface area. Stomatocytosis has been reported in alcoholic liver disease.
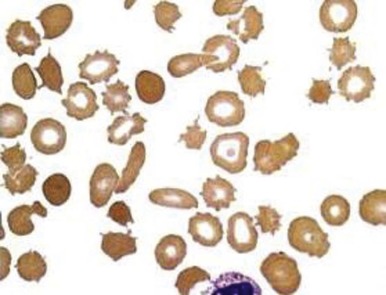
Coagulation Abnormalities
Because the liver is involved to some extent in the synthesis of most of the coagulation factors, it is not unexpected that liver dysfunction would be associated with the presence of abnormal clotting studies and bleeding. The coagulation abnormalities common in patients with fulminant hepatic failure have been reviewed in detail.
Patients with liver disease commonly manifest a spectrum of coagulation abnormalities that are highly suggestive of DIC. Findings consistent with DIC include hypofibrinogenemia, thrombocytopenia, increased fibrinogen catabolism, increased levels of fibrin degradation products, and depressed levels of other coagulation factors. This pattern of abnormalities is not specific for DIC and may simply reflect the severity of the hepatocellular disease process. For example, fibrinogen levels may be depressed on a synthetic basis alone. Increased catabolism of fibrinogen may reflect distribution in extravascular spaces, such as the formation of ascitic fluid or proteolysis by enzymes other than thrombin. Thrombocytopenia may occur for a variety of reasons and frequently is not present when other signs of DIC are seen. Changes in levels of factors V and VIII are varied and nonspecific. Because fibrin degradation products are cleared by the liver, severe liver dysfunction itself may cause elevations of these products without implicating DIC.
The acquired coagulation abnormalities of liver disease are discussed in detail in Chapter 35 .
Endocrine Disorders
Disturbances in endocrine balance tend to produce relatively mild hematologic abnormalities. The most thoroughly evaluated of these abnormalities are those caused by thyroid disorders.
Thyroid Gland
Thyroid hormone appears to have many hematologic effects ( Table 37-4 ), with the most impressive being its effect on erythropoiesis. Individuals with hyperthyroidism sometimes have an elevated red blood cell mass secondary to the effects of excessive thyroid hormone and have an increased requirement for folic acid to accommodate the increased red blood cell synthesis. The red blood cells of patients with hyperthyroidism may have increased osmotic resistance. The red blood cell sodium concentration is significantly increased in hyperthyroidism, and decreased sodium pump activity may be present. A macrocytic anemia may complicate hyperthyroidism in the setting of Graves disease because pernicious anemia and other immune disorders are linked to Graves disease. Neutropenia may occur in approximately 5% of children with hyperthyroidism. The mechanism of this abnormality is unclear, although some patients will have antineutrophil antibodies.
| ↑ Thyroid Hormone | ↓ Thyroid Hormone |
|---|---|
| ↑ PRBC volume | ↓ PRBC volume |
| Variable effect on RBCs | ↑ Or normal MCV; spiculated RBCs |
| ↑ Glucose utilization and hexose monophosphate shunt activity | ↑ O 2 affinity |
| ↓ RBC glutathione, reduced | ↓ 2,3-Diphosphoglycerate |
| ↑ Red blood cell 2,3-diphosphoglycerate | ↓ RBC Na + |
| ↓ O 2 affinity | ↓ Platelet adhesion |
| ↑ Diphosphoglycerate mutase activity | ↓ (Slight) platelet aggregability |
| ↑ Glyceraldehyde-3-phosphate dehydrogenase | ↓ Factor VIII activity |
| ↑ Glucose 6-phosphate activity | ↑ Fibrinolytic activity |
| ↓ RBC carbonic anhydrase | ↑ Plasminogen |
| ↓ RBC zinc | ↑ Plasminogen activation |
| ↑ RBC Na 2+ | |
| ↑ (Slight) platelet turnover | ↓ Capillary fragility |
| ↑ Platelet adhesion | ↑ Factors VIII, VII, IX, XI activity |
| ↑ Platelet aggregability | |
| ↑ Factor VIII activity | |
| ↓ Plasminogen activation | |
| ↑ Capillary fragility |
The anemia of hypothyroidism is caused by a reduced red blood cell mass secondary to the decreased oxygen requirements of the hypothyroid state. The anemia is usually normochromic and normocytic and occasionally mildly macrocytic. Patients with hypothyroidism often (20% to 65%) have a small number (0.5% to 2%) of acanthocytes on peripheral blood smears. Given the high incidence of hypothyroidism relative to other disorders that cause acanthocytosis, it has been suggested that the presence of acanthocytes should prompt physicians to consider thyroid testing, especially in adults. This approach has led to the diagnosis of previously unsuspected hypothyroidism. Red blood cell survival is normal, and ferrokinetic studies demonstrate a decreased rate of iron clearance. In adults with hypothyroidism, an unusually high incidence of iron deficiency, often linked to menorrhagia, and pernicious anemia is found.
Thyroid hormone may play a role in the regulation of red blood cell metabolism. Thyroxine has been reported to increase red blood cell glycolysis. It helps regulate red blood cell carbonic anhydrase I concentrations. Hexose monophosphate shunt activity may increase in the presence of triiodothyronine. The activity of glucose-6-phosphate dehydrogenase is consistently elevated in thyrotoxicosis, whereas normal levels are found in most patients with mild hyperthyroidism. Glutathione levels are usually depressed in hyperthyroidism. Thyroid hormone may affect the activities of several other red blood cell enzymes. This hormone stimulates diphosphoglycerate mutase and glyceraldehyde-3-phosphate dehydrogenase. These effects may be the cause of the elevated levels of red blood cell 2,3-DPG that are seen in patients with hyperthyroidism and that result in decreased whole blood oxygen affinity and a shift to the right in the hemoglobin-oxygen dissociation curve. Opposite effects are seen in hypothyroidism. The carbonic anhydrase activity of erythrocytes is diminished in hyperthyroidism and increased in hypothyroidism. Because red blood cell zinc is almost exclusively contained in carbonic anhydrase, hyperthyroidism will result in a decrease in total red blood cell zinc levels.
Multiple effects on platelets and blood coagulation factors may occur in thyroid disease states. Marked hyperthyroidism may reduce platelet survival. Egeberg reported on a patient with hypothyroidism who had a prolonged bleeding time and low factor VIII activity, similar to findings in von Willebrand disease.
Simone and associates reported on patients with hypothyroidism who had multiple coagulation factor deficiencies. Diminished platelet responsiveness to epinephrine and low platelet adhesiveness may be present as well. Impressively low levels of factor VIII occur occasionally, with milder depression of factors VII, IX, and XI being found either alone or in combination. In hypothyroidism, increased plasma fibrinolytic activity may result from an elevation in plasma plasminogen and a decrease in an inhibitor of plasminogen activation. This may account for the mild to moderate increase in levels of fibrinogen and fibrin split products occasionally found in patients with hypothyroidism.
Hypothyroidism is also associated with acquired type I von Willebrand disease in 2% to 5% of cases. The low von Willebrand factor antigen is due to a reduction in its synthesis and/or secretion. Similar to inherited type I von Willebrand disease, acquired von Willebrand disease in hypothyroidism is associated with mucocutaneous bleeding and responds to desmopressin and antifibrinolytic agents. Acquired von Willebrand disease typically reverses with correction to a euthyroid state.
Adrenal Gland
Adrenal corticosteroids appear to have a stimulatory effect on erythropoiesis. In Addison disease, the adrenal insufficiency may cause mild to moderate anemia, probably as a result of reduced basal metabolism. In most patients, a decrease in plasma volume masks the decreased red blood cell mass. After treatment of adrenal insufficiency, the plasma volume corrects quickly, whereas the red blood cell mass responds over a period of several weeks. The usual effect of treatment is therefore a prompt decrease in hemoglobin concentration followed by a gradual increase. In addition to anemia in Addison disease, neutropenia, eosinophilia, and lymphocytosis are common.
Erythrocytosis may occur in Cushing syndrome, but in most patients the reported increase has been slight. Cushing syndrome is also associated with coagulation abnormalities, suggesting a hypercoagulable state.
A variety of effects on leukocytes occur after corticosteroid administration or endogenous overproduction of a corticosteroid, including granulocytosis, a reduced lymphocyte count, involution of lymphatic tissue, and a decrease in peripheral blood eosinophils and monocytes. The mechanism by which endogenous or exogenous corticosteroids lower the number of circulating eosinophils is unknown.
Pituitary Gland
The hematologic effects of pituitary disease are usually a consequence of the action of trophic hormone on target endocrine organ function. For example, anemia is common in patients with hypopituitarism. This normochromic, normocytic anemia is associated with findings of lymphocytosis and eosinophilia, which indicate that this effect of hypopituitarism results from adrenal insufficiency. Erythropoietin production may be diminished as a consequence of a lower metabolic rate.
Growth hormone may play an important role in erythropoiesis. This hormone stimulates erythropoiesis directly and indirectly by increased production of erythropoietin. The red blood cell count may decrease with isolated growth hormone deficiency. Erythrocyte glucose-6-phosphate dehydrogenase activity is low in hypopituitarism and increases with growth hormone administration. Growth hormone depresses erythrocyte glycolysis.
Other Endocrine Disorders
Hematologic abnormalities are rare in association with gonadal dysfunction. Androgens stimulate erythropoiesis, whereas estrogens, in general, depress red blood cell production. Castration of the adult male results in a definite decrease in red blood cell mass. In disorders associated with androgen excess, such as Cushing syndrome and congenital adrenal hyperplasia, the hemoglobin concentration may exceed normal values. Exogenous androgen administration may cause polycythemia and may also increase red blood cell 2,3-DPG. Androgens appear to stimulate erythropoiesis by increasing erythropoietin production, as well as by having a direct effect on bone marrow stem cells.
Diabetes Mellitus
It has long been recognized that diabetic patients are prone to anemia, infection, and thrombotic episodes with macrovascular and microvascular sequelae.
Red Blood Cells
An unusual hemoglobin component in hemolysates prepared from the blood of some diabetic patients was first noted by Rahbar. A component that migrated near the position of fetal hemoglobin was described on agar gel electrophoresis at pH 6.2 in citrate buffers. This hemoglobin is present in normal individuals; it constitutes 5% to 7% of total hemoglobin and is called hemoglobin A 1c . Rahbar’s group observed a twofold increase in this hemoglobin fraction in some diabetic patients. Structurally, hemoglobin A 1c is a condensation product, by means of a Schiff base, between one molecule of hemoglobin A and one molecule of an aldehyde or ketone, linked at the amino-terminals. The aldehyde or ketone group appears to be one or more hexoses, thus making hemoglobin A 1c a glycohemoglobin. Bunn and Briehl have shown that the oxygen affinity of hemoglobin A 1c is little affected by the addition of 2,3-DPG, which leads to decreased oxygen affinity when added to hemoglobin A. Studies have shown highly significant relationships between the percentage of hemoglobin A 1c and the response to an oral glucose tolerance test and overall diabetic control, as reflected in quantitative urinary glucose determinations. In this sense, measurement of the glycohemoglobin provides an insight into diabetic control over many days.
Red blood cell survival, as measured by chromium ( Cr) labeling, may be mildly impaired during periods of poor diabetic control (mean erythrocyte half-life of 27 days). With improvement of diabetic control, red blood cell survival will increase (mean half-life of 31 days). During episodes of ketoacidosis, rapid shifts in the oxygen affinity of hemoglobin may also occur. A decrease in pH causes a shift to the right with an increase in oxygen unloading. Very rapid correction of pH may impede oxygen delivery. A prompt decrease in red blood cell 2,3-DPG levels occurs at these times. This decrease is probably compensatory—an attempt to correct the oxygen affinity disturbances caused by the acidosis. A decrease in red blood cell 2,3-DPG levels may also result from hypophosphatemia, if it occurs during insulin treatment.
Other red blood cell abnormalities have been found in diabetic subjects. The red blood cells of such individuals show evidence of lipid peroxidation and antiperoxidative enzyme changes when compared with those of healthy subjects. An increase in the glycosylated form of erythrocyte superoxide dismutase has been found in diabetic subjects and is related to nonenzymatic glycosylation of this enzyme. Erythrocytes from diabetic subjects also show sodium influx. Red blood cell sorbitol concentrations will increase in subjects with poorly controlled diabetes; the amount of red blood cell sorbitol is related to the degree of diabetic control. Finally, if electronic counting equipment is used to determine the mean corpuscular volume of red blood cells, artifactual elevations in mean corpuscular volume may be seen in subjects with hyperglycemia.
Thiamine-responsive megaloblastic anemia, diabetes mellitus, and sensorineural deafness is a rare autosomal recessive disorder that begins in childhood and is caused by defects in the high-affinity thiamine transporter-1 (SLC19A2), on chromosome 1q23.3. The anemia and diabetes respond to high-dose thiamine treatment, although the diabetes can relapse years later, despite continued thiamine treatment.
White Blood Cells
Polymorphonuclear cell function may be disturbed in diabetic patients. Leukocyte adherence is diminished in individuals with poor control of diabetes, as is phagocytic capacity. Chemotaxis may also be abnormal, but this does not correlate with the status of diabetic control. Impaired cellular metabolism and DNA synthesis have been reported in the lymphocytes of some patients with diabetes. Increased superoxide production by mononuclear cells is observed in patients with diabetes and hypertriglyceridemia.
Platelets
Many studies have emphasized the abnormalities in platelet adhesion and aggregation in patients with diabetes. The finding that enhanced platelet aggregation occurs before clinical evidence of diabetic vascular disease suggested that this defect may be acquired early in the natural history of diabetes and could underlie the vascular disease. Platelet aggregation in diabetic subjects is characterized by a shortened adenosine diphosphate–induced aggregation time. Increased platelet adhesiveness and platelet factor 3 and 4 activity also occur. Normal platelets incubated in the plasma of diabetic patients will become abnormal. This aggregation-enhancing activity is present in both plasma and serum and is nondialyzable and heat resistant. In patients with this plasma factor, von Willebrand factor activity is also increased, thus suggesting that the two are related. The enhanced in vitro responsiveness of platelets from diabetic patients can be decreased by prostaglandin synthetase inhibitors. Platelets from diabetic patients demonstrate increased activity of the prostaglandin synthetase system, which results in increased synthesis of prostaglandin endoperoxides and, therefore, of prostaglandin E 2 . When induced to aggregate with arachidonic acid, the platelets of diabetic patients with vascular disease tend to produce more thromboxane A 2 than those of normal subjects do. A lower prevalence and severity of diabetic retinopathy have been observed in a group of diabetic patients with concurrent rheumatoid arthritis who were taking high doses of aspirin. This finding suggests that inhibition of platelet aggregation and prostaglandin synthesis may be desirable in the management of diabetic patients. The increased glycosylation of connective tissue proteins in diabetic patients has been suggested to increase their aggregation potency.
Other Hematologic Findings in Diabetes Mellitus
A state of hypercoagulability associated with changes in clotting factors and platelet function has been postulated to be important in the increased thrombotic complications in diabetic patients. Approximately a third of patients with diabetes will show abnormally increased factor VIII coagulant activity and diminished antithrombin III levels. Abnormalities in fibrinolysis do not appear to be common. In addition to this, factor XI and factor XII levels are higher in some diabetic patients with evidence that the kallikrein-prekallikrein system has been activated. Ketoacidosis may be associated with DIC. Altered levels of factors V and VIII and diminished fibrinolytic activity have been reported in a few patients.
The hematologic consequences of diabetes also extend to infants born of diabetic mothers. An increased incidence of thrombosis or thromboembolic phenomena is well recognized in these infants. Complications such as renal vein thrombosis, peripheral gangrene secondary to vascular occlusion, and cerebral thrombosis may occur.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


