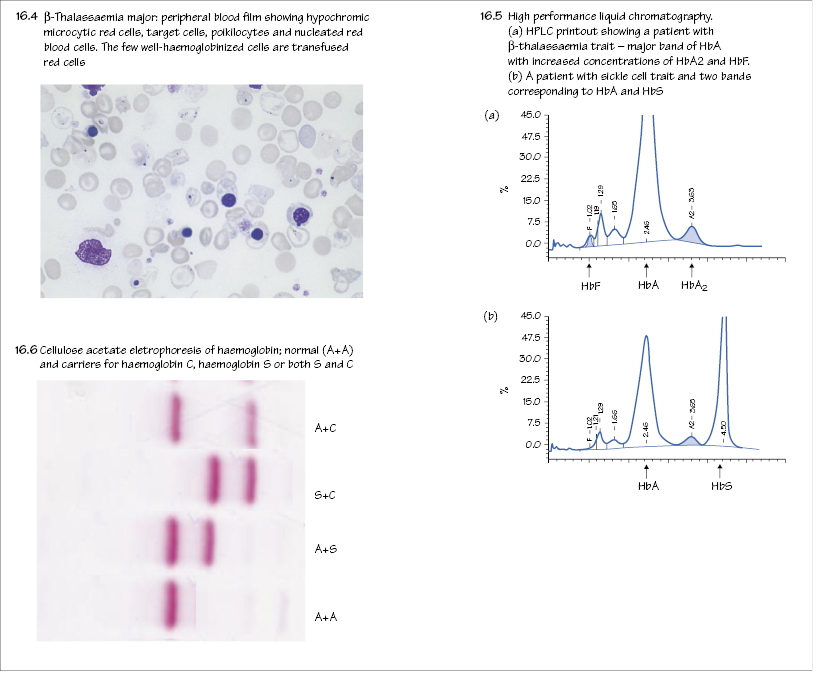
Normal haemoglobins
The haemoglobins are made up of four globin chains each containing a haem group. Embryonic haemoglobins (Portland, Gower I and II) are present in early fetal life. Fetal haemoglobin (Hb F, α2γ2¬) dominates by late fetal life. Hb F has a higher oxygen affinity than Hb A and this allows the fetus to obtain oxygen from the mother (Fig. 2.3). A switch occurs at 3–6 months in the neonatal period to normal adult haemoglobin (Hb A, α2δ2) (Fig. 16.1). Low levels of Hb F and the minor adult haemoglobin Hb A2 (α2δ2) are also present in normal adults (see Table 2.1).
Genetic disorders of haemoglobin
Table 16.1 Classification of thalassaemia
| Clinical phenotype | Thalassaemia (thal) syndrome |
|---|---|
| Hydrops foetalis | Homozygous α-thal major → complete lack of α-globin |
| Thalassaemia major | Homozygous β or doubly heterozygous β thal → complete or almost complete lack of β-globin |
| Thalassaemia intermedia | See below |
| Thalassaemia trait | Heterozygous β-thalassaemia (β-thal minor, lack of one functional β-globin gene*) |
| Heterozygous α-thalassaemia (α-thal minor, lack of one or two α-globin genes†) |
*Normal individual has two (one from each parent on each allele)
†Normal individual has four (two from each parent on each allele)
The first and second have a wide global prevalence, particularly where malaria is, or was, common, as the carrier states give some protection against falciparum malaria. Compound heterozygote states of a thalassaemic allele and a haemoglobin structural variant allele frequently occur and include sickle/β-thalassaemia and Hb E/β-thalassaemia.
Thalassaemia
These autosomal recessive syndromes divide into α- and β-thalassaemia depending on whether there is reduced synthesis of α- or β-globin (Table 16.1, Fig. 16.2).
α-Thalassaemia
Normally there are four α-globin genes, two on each chromosome 16 (Fig. 16.1). The severity of α-thalassaemia depends on the number of α-genes deleted or, less frequently, dysfunctional.
Hydrops foetalis
Related posts:
 Clinical assessment
Megaloblastic anaemia II: clinical features, treatment and other macrocytic anaemias
Haemolytic anaemias V: genetic defects of haemoglobin – sickle cell disease
Lymphoma II: Hodgkin lymphoma
Stem cell transplantation
Clinical assessment
Megaloblastic anaemia II: clinical features, treatment and other macrocytic anaemias
Haemolytic anaemias V: genetic defects of haemoglobin – sickle cell disease
Lymphoma II: Hodgkin lymphoma
Stem cell transplantation
 Disorders of haemostasis II: inherited disorders of coagulation
Disorders of haemostasis II: inherited disorders of coagulation
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


