apoptosis at peripheral sites—would consist in a decrease in the levels of neutrophils released from the bone marrow.7 On the other hand, the number of circulating neutrophils can dramatically increase (even up to 10-fold) under acute inflammatory conditions (eg, during a bacterial infection), from accelerated neutrophil production and release from the bone marrow.4 Moreover, even the lifespan of neutrophils is significantly extended under inflammatory conditions, as various host- and pathogen-derived mediators such as G-CSF, GM-CSF, interferon (IFN)-γ, tumor necrosis factor (TNF)-α, lipopolysaccharide, and nucleic acids inhibit neutrophil apoptosis and hence prolong their survival.8
 FIG. 20.1. A Polymorphonuclear Neutrophil Circulating in Peripheral Blood. From Anderson’s Atlas of Hematology; Anderson, Shauna C., PhD. Copyright 2003, Wolters Kluwer Health/Lippincott Williams & Wilkins. |
major weapons against microbes, as it also synergizes with granule proteins to kill pathogens in the neutrophil phagolysosome. O2– can also reacts with other cellular radicals, such as nitric oxide, to form different species of cytotoxic oxidant, such as peroxynitrite. The critical role of NADPH oxidase and its products in host defense is best illustrated by the plight of patients with chronic granulomatous disease, in which mutations in any of the NADPH oxidase complex subunits (gp91phox, p22phox, p40phox, p47phox, and p67phox) leads to a severe immunodeficiency characterized by defective killing of phagocytosed pathogens for the lack of ROS generation.10 These infections typically involve microorganisms for which oxidant-mediated killing is particularly critical for effective host defense, such as Staphylococcus aureus, Aspergillus spp., Nocardia, and a variety of gramnegative enteric bacilli.
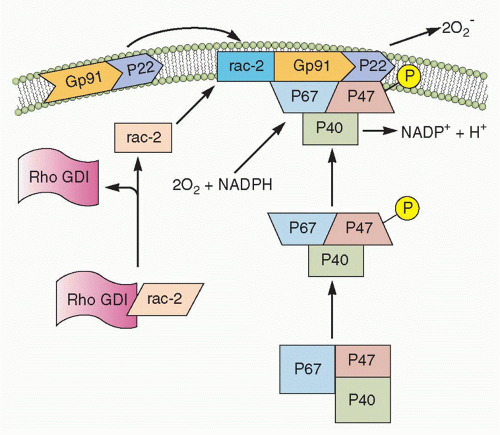 FIG. 20.2. Nicotinamide Adenine Dinucleotide Phosphate (NADPH) Oxidase Assembly. In the resting neutrophil, the cytochrome b subunits gp91-phox and p22-phox are tightly bound in the membrane. p47-phox, p67-phox, and rac-s complex are in the cytosol. Upon activation, Rho GDP-dissociation inhibitor (GDI) releases rac-2, and p47-phox becomes phosphorylated. This causes translocation of rac-2, p47-phox, and p67-phox to the membrane and complex formation with the cytochrome components, thereby completing the assembly of the active oxidase. (After Burg ND, Pillinger MH. Clin Immunol. 2001;1:7-17.) |
of nuclear neutrophil segmentation in the bone marrow, and are the most readily mobilizable.11,12 These vesicles are preferentially directed to the plasma membrane, as reflected in the density of vesicle-associated membrane protein (VAMP), a fusogenic protein associated with the granule membrane. Secretory vesicles do not contain toxic substances, but mainly plasma proteins like albumin and receptors (including β2-integrins, the complement receptor [CR]1, receptors for formylated bacterial peptides [fMLF-R], CD14, the Fc portion of γ-immunoglobulins (Igs) [FcγRIII/CD16], and the metalloprotease leukolysin). Heparin-binding protein (also known as CAP37 or azurocidin), whose release is essential for the polymorphonuclear leukocyte-induced increase in vascular permeability at the initial stage of extravasation, is also stored in the secretory vesicles.
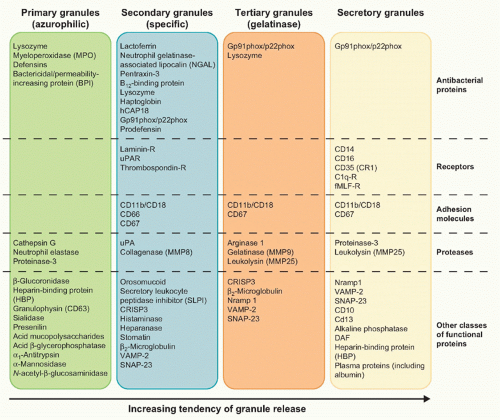 FIG. 20.3. Main Constituents of Neutrophil Granules. |
and group A streptococci), as well as to pathogenic fungi (such as Candida albicans). Similarly to what happens in the phagolysosome, the high local concentration of antimicrobial peptides and enzymes is responsible for the killing of the pathogens trapped by NETs.13 The observation that neutrophils from patients with chronic granulomatous disease do not form NETs has suggested, on the one hand, that ROS-mediated signaling/cascades are involved in NET generation, and, on the other hand, that the lack of NETs might contribute to the pathogenesis of chronic granulomatous disease.3,14 Whatever the case, it is noteworthy to remark that both the oxygen-dependent and -independent effector mechanisms in host defense toward pathogens are also utilized by neutrophils for their cytotoxic and tumoricidal activities.
injury or infection and, once arrived, begin to react with the etiopathogenic agent.
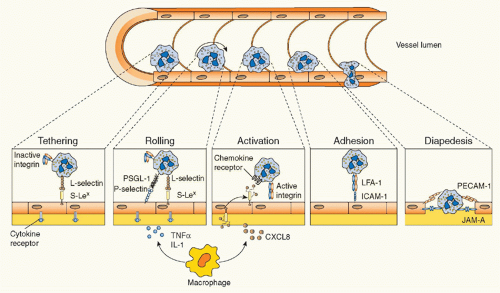 FIG. 20.4. Leukocyte Transmigration. Following an inflammatory stimulus, tissue-resident macrophages and other cells release inflammatory mediators such as tumor necrosis factor α and interleukin-1, which induce the rapid expression of preformed P-selectin (and transcription-dependent E-selectin expression) on the endothelium. The interaction between selectins and their glycoprotein ligands initiates leukocyte tethering and rolling. Activation by chemokines—and other leukocyte activators (eg, leukotriene-B4 or platelet-activating factor)—presented on endothelial cells causes leukocyte integrin activation, thus resulting in transition from cell rolling to cell firm adhesion, in view of the strength of integrin-mediated binding with endothelial immunoglobulin superfamily members. Leukocytes can then transmigrate through the endothelial monolayer and chemotactically move toward the inflammatory stimulus. Examples of adhesion molecules involved in each step are depicted. |
FcγRs (FcγRIIA/CD32A and FcγRIIIA/CD16A), and, when exposed to IFNγ or G-CSF, the high-affinity FcγR (FcγRI/CD64) as well.3 CRs expressed by neutrophils are CR1 (also known as CD35), which binds to complement components C1q, C4b, C3b, and mannan-binding lectin; CR3, which binds to iC3b, intercellular adhesion molecule-1, and some microbes; and CR4, which binds to iC3b. By expressing these latter receptors, neutrophils are able to recognize and bind, in a cooperative manner, IgG-opsonized particles and/or complement-opsonized microbes, and then activate their phagocytosis. During the phagocytic process, the foreign particle is internalized, initially through membrane recruitment to the site of particle contact, and then via membrane extensions outward to surround the particle and form a new vesicle called a cytoplasmic phagosome.20 (Fig. 20.5). The phagosome then undergoes fusion with neutrophil granules to form a phagolysosome, a protected space in which proteolytic enzymes and other bactericidal components are discharged and pathogen degradation occurs. At the same time, NADPH oxidase assembles on the phagosomal membrane after phagocytosis and starts to generate ROS into the phagolysosome to kill bacteria by oxidizing microbial proteins and lipids. The activity of NADPH oxidase also leads to the acidification of the phagosome, which enhances the effectiveness of pH-sensitive antimicrobial compounds. Thus, neutrophil mechanisms of pathogen destruction within the phagosome are multiple and involve granule fusion, toxic oxygen radical production, activation of latent proteolytic enzymes, and the activity of antibacterial proteins (see Fig. 20.5). Remarkably, an activation of gene transcription and a selected generation of cytokines also occur during phagocytosis, a feature that neutrophils utilize for boosting a more effective innate immune response. For instance, recruited neutrophils that phagocytose
a pathogen also respond by producing chemokines, in particular CXCL8, to amplify their own recruitment, but also CCL3, CCL4, and CCL19 that serve to recruit monocytes and DCs.21
TABLE 20.1 Main Adhesion Molecules Involved in Leukocyte-Endothelial Cell Interaction | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
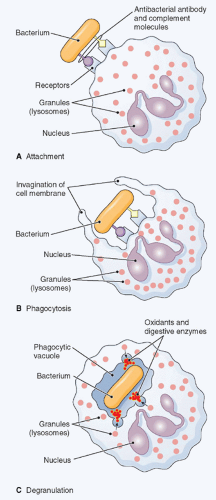 FIG. 20.5. Phagocytosis. The figure shows ingestion, digestion, and destruction of foreign particulate matter (a bacterium, in this example) by a neutrophil. A: Cell membrane receptors bind to antibody and complement molecules previously attached to the bacterial surface. B: The cell membrane creeps around the bacterium and envelopes it. C: The bacterium is trapped in a special space, the phagocytic vacuole, into which lysosomes discharge proteases, which together with oxidants kill it. Then, digestive enzymes dissolve it. (Thomas H. McConnell, The Nature Of Disease Pathology for the Health Professions, Philadelphia: Lippincott Williams & Wilkins, 2007.) |
basis, fewer molecules of a given cytokine than mononuclear leukocytes.21 However, considering that neutrophils clearly predominate over other cell types under inflammatory conditions in vivo, it becomes obvious that the contribution of neutrophil-derived cytokines can be of foremost importance. To date, a wide range of stimuli able to induce characteristic signatures of chemokine and cytokine synthesis by neutrophils have been identified. Among these, cytokines themselves, chemotactic factors (fMLF, LTB4, PAF, C5a, and CXCL8), phagocytic particles, microorganisms (such as fungi, viruses, and bacteria), and PRR ligands can all induce the synthesis and release of chemokines and cytokines by neutrophils.21 Considering that neutrophils usually represent the first cell type infiltrating at the site of infections, a stimulus-specific response of neutrophils in terms of cytokine production might direct the evolution of certain types of inflammatory and immune reactions to support the transition from innate to adaptive immunity.
TABLE 20.2 Cytokines Expressed in Resting or Activated Neutrophils | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||
family that are well known to be essential for B-lymphocyte homeostasis.25 Therefore, it is plausible to assume a role of neutrophils not only in sustaining B and plasma cell antibody production and survival, but also in promoting B-cell-dependent autoimmune diseases and tumors, as already elegantly demonstrated in the case of B-cell lymphoma.25
neutrophils of all these immunosuppressive granulocytic populations. Such studies will better clarify the real role of neutrophils in cancer as well as in other inflammatory/autoimmune diseases (such as infections, psoriasis, and lupus), in which the presence of MDSC- or low-density granulocyte -like cells have been also described.40
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


