Chapter Outline
G6PD IN RED BLOOD CELL METABOLISM
Structure and Biochemistry of G6PD
Features of G6PD in Red Blood Cells
Molecular Basis of G6PD Deficiency
CLINICAL MANIFESTATIONS OF G6PD DEFICIENCY
LABORATORY DIAGNOSIS OF G6PD DEFICIENCY
GENOTYPE-PHENOTYPE CORRELATIONS
G6PD DEFICIENCY AND PREVENTIVE MEDICINE
G6PD DEFICIENCY IN NONERYTHROID CELLS
G6PD DEFICIENCY COEXISTING WITH OTHER DISORDERS
Most hemolytic anemias can be categorized, at least at first approximation, as being either inherited or acquired as a result of either intracorpuscular or extracorpuscular causes. Hemolytic anemia associated with deficiency of glucose-6-phosphate dehydrogenase (G6PD) glaringly defies this categorization. Indeed, most persons with inherited G6PD deficiency have no anemia and almost no hemolysis; both develop only as a result of challenge by exogenous agents. Because the metabolic role of G6PD in red blood cells is primarily related to its reductive potential, the threat to G6PD-deficient red blood cells is oxidative damage. Therefore to understand hemolytic anemia associated with G6PD deficiency, we first need to define the physiologic role of G6PD and pinpoint why the red blood cells are deficient in G6PD.
G6PD in Red Blood Cell Metabolism
In metabolic maps, G6PD is commonly referred to as the first enzyme of the hexose monophosphate shunt, or the pentose phosphate pathway ( Fig. 18-1 ). Although these time-honored phrases persist in textbooks, it is now clear that the main role of G6PD is not glucose utilization (G6PD accounts for less than 10% of that function); rather, its main role is production of the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH). NADPH has a crucial role in preventing oxidative damage to proteins and other molecules in all cells ( Fig. 18-2 ). This role is particularly crucial in red blood cells because, being oxygen carriers par excellence, they have a literally built-in danger of damage by oxygen radicals generated continuously in the course of methemoglobin formation. The highly reactive oxygen radicals either decay spontaneously or are converted by superoxide dismutase to hydrogen peroxide (H 2 O 2 ), which is still highly toxic. Detoxification of H 2 O 2 to H 2 O is effected by catalase and glutathione peroxidase (GSHPX). NADPH is crucial for the function of both these enzymes; it is a structural component of catalase and is required as a substrate by glutathione reductase, which regenerates reduced glutathione (GSH) when it has been oxidized to oxidized glutathione by GSHPX (see Fig. 18-2 ). G6PD-deficient red blood cells are highly vulnerable to oxidative damage (discussed later in this chapter), even though G6PD deficiency is never complete in humans. When complete G6PD deficiency was produced in mouse embryonic stem cells by targeted inactivation of the G6PD gene, the G6PD-null cells thus obtained were viable, but they formed colonies only in a low-oxygen environment; even so, they had an impaired capacity to form erythroid colonies. When G6PD-null embryonic stem cells were injected into mouse blastocysts, chimeric embryos were obtained, and germline transmission was achieved; however, only female heterozygous mice were obtained because hemizygous male embryos died. Thus a G6PD-null mutation is an embryonically lethal condition.
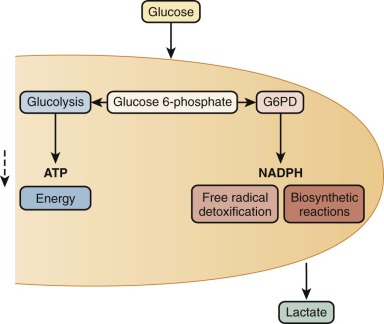
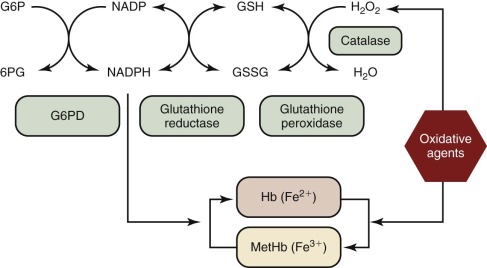
Structure and Biochemistry of G6PD
G6PD is a ubiquitous enzyme that must be quite ancient in evolution because it has been found in all organisms, from prokaryotes to yeasts, protozoa, plants, and animals. In mammals, G6PD is a good example of a housekeeping enzyme produced by a housekeeping gene; indeed, the gene is expressed in all cells of the body. G6PD is a typical cytoplasmic enzyme, although some G6PD activity is associated with peroxisomes in liver and kidney cells. This finding is consistent with the view that these organelles have evolved as part of the need of early eukaryotes to defend against oxygen, which is germane to the role of G6PD today.
The enzymatically active form of G6PD is either a dimer or a tetramer ( Fig. 18-3 ) of a single polypeptide subunit of about 59 kD. The complete primary structure of the human enzyme has been deduced from the sequence of a full-length complementary deoxyribonucleic acid (DNA) clone. The amino acid sequence of rat liver G6PD shows 94% homology to the human sequence and provides evidence that the N-terminal amino acid is N -acetylalanine, which must result from posttranslational cleavage of the N-terminal methionine. The same is probably true of the human enzyme.
Extensive data are available on the kinetics of G6PD. Its coenzyme specificity is exquisite; human G6PD has practically no activity with nicotinamide adenine dinucleotide. Its substrate specificity is also very high because activity on other hexose phosphates (e.g., mannose 6-phosphate or galactose 6-phosphate) is negligible. The affinity for nicotinamide adenine dinucleotide phosphate (NADP) is about one order of magnitude higher than the affinity for glucose 6-phosphate (G6P).
In the three-dimensional structure of G6PD (see Fig. 18-3 ), one sees that the G6P binding site is near K205, and the critical role of this amino acid in electron transfer has been confirmed by showing that replacing it with threonine nearly abolishes catalytic activity. The NADP binding site is located near a fan of β-sheet structures, with a critical G-X-X-G-X-X peptide motif corresponding to amino acids 38 to 43 encoded in exon 3. Interestingly, the presence of a molecule of “structural” NADP, which had been suggested by early biochemical work, has been confirmed.
Although many natural and nonnatural substances can affect the activity of G6PD, it is not certain which ones may be important physiologically. NADPH, one product of the G6PD reaction, is a potent quasicompetitive inhibitor, and because most of the coenzyme in cells is in the reduced form, it can be assumed that G6PD is normally under strong inhibition. Because the normal intracellular concentrations of both G6P and NADP are lower than their respective Michaelis constant ( K m ) values, it is likely that these two substrates themselves are the main regulators of intracellular G6PD activity, together with NADPH. Any oxidative event affecting the cell will alter the NADPH/NADP ratio in favor of NADP. The simultaneous increase in NADP and decrease in NADPH act additively to increase G6PD activity by increasing the substrate drive on the reaction rate and decreasing product inhibition. Under most conditions this may be the most important short-term regulatory signal, although it is, of course, possible that other regulatory effects play a role as well. Recently, as part of a study of the role of microribonucleic acids in Duchenne muscular dystrophy myoblasts, it has been discovered that miR-1 is a powerful negative regulator of G6PD synthesis. Although not relevant to mature red blood cells, this phenomenon may be important in determining the G6PD level in nucleated cells both physiologically and under stress conditions.
Notes on Terminology
G6PD is the accepted abbreviation for the enzyme glucose-6-phosphate dehydrogenase (E.C. 1.1.1.49); the G6PD gene is designated G6PD or Gd . Because Gd is X-linked (discussed later in this chapter), males can be only normal hemizygotes ( Gd +) or deficient hemizygotes ( Gd −); females can be normal homozygotes ( Gd +/ Gd +), deficient homozygotes ( Gd −/ Gd −), or heterozygotes ( Gd +/ Gd −). The phenotype of the last group is often referred to as “intermediate” because their overall red blood cell G6PD level usually lies in between the normal and the deficient range; however, exceptions do occur and will be discussed later in this chapter. Consequently, G6PD deficiency should not be regarded as a recessive but rather as a codominant trait. In this chapter, by G6PD deficient, we mean Gd − males and Gd − / Gd − females; Gd + / Gd − females will have some degree of G6PD deficiency, but for clarity, we will always specify heterozygotes. According to a classification introduced in 1966 ( Table 18-1 ), G6PD-deficient variants that result in congenital nonspherocytic hemolytic anemia (CNSHA) are designated class I; G6PD-deficient variants that do not result in CNSHA are designated class II or class III, depending on the severity of the reduction in enzyme activity in red blood cells. The separation between class II and class III is blurred and probably no longer useful. Class IV variants are those with normal activity. Class V was reserved for variants with increased activity, but after an initial report (G6PD Hektoen ), none has been found. In practice, because the majority of G6PD-deficient persons are mostly asymptomatic, their G6PD deficiency is referred to as mild, simple, or common (corresponding to class II or III); the minority of persons who have CNSHA are referred to as having rare, sporadic, or severe G6PD deficiency (corresponding to class I).
| Class * | Clinical Manifestations † | G6PD Activity (% of Normal) | No. of Known Mutant Alleles ‡ | Examples | Comments |
|---|---|---|---|---|---|
| IV | None | >85 | 2 | A,B | G6PD B is the normal “wild type” |
| II + III | Asymptomatic in the steady state, but risk for NNJ, AHA, favism | <30 | 75 | Med, A − , Orissa, Mahidol, Canton Vanua Lava, Seattle | Most of these variants are known to be polymorphic |
| I | NNJ (severe), CNSHA | <10 in most cases | 61 | Sunderland, Nara, Guadalajara | Never polymorphic, but the same mutation can recur |
* The separation between class II and class III is blurred and, in our opinion, is no longer useful.
† In brief, variants in class II and III can be regarded as having a mild phenotype confined to acute episodes; variants in class I have a severe phenotype with chronic illness.
‡ See Fig. 18-4, B . We have counted only variants for which the mutation is known and the clinical expression is clearly reported.
When a diagnostic test for G6PD is carried out, the phrase “positive result” is sometimes used to indicate that the test has shown G6PD deficiency. Use of this phrase has caused confusion, and its use should be avoided; instead, one should indicate that the test has shown either a normal result or a deficient result.
Genetics of G6PD
Gd , the G6PD gene, is located near the telomeric region of the long arm of the X chromosome (band Xq28); historically, Gd has been a valuable X-linked genetic marker and a precious tool for studying the X-chromosome inactivation phenomenon and clonal populations of somatic cells. The Gd region was also one of the first regions of the human genome to be fully sequenced.
X-linkage of Gd has three major consequences: (1) Gd mutations display the typical pattern of mendelian X-linked inheritance ; (2) severe G6PD deficiency is much more common in males than in females; and (3) as a result of X-chromosome inactivation, females who are heterozygous for two different Gd alleles exhibit somatic cell mosaicism. As a result, if one of the alleles entails enzyme deficiency, about half the cells will be G6PD(+) and the other half will be G6PD(−), although there is a large range of variation around that average for various reasons, not all of them fully known. At first approximation it can be assumed that X-inactivation takes place at random, and therefore a binomial distribution would be expected; the width of this distribution depends on the number of cells in the embryo or in individual embryonic tissue at the time of X-inactivation. If this number is 32 to 64, a fraction of about 2% of women with an “extreme phenotype” is predicted, that is, with less than 5% of one of the two cell types; this prediction is in good agreement with observation in an unselected sample of Gd +/ Gd − heterozygotes, although some studies have suggested an even larger proportion.
Thus in many cases unbalanced or skewed phenotypes may arise simply by chance, according to the laws of statistics. However, in certain cases evidence exists of selection at the somatic cell level after X-chromosome inactivation. This finding was first well established in women who are heterozygous for hypoxanthine phosphoribosyltransferase deficiency, and it has also been observed in several women heterozygous for severely deficient G6PD variants. In these cases one must infer that the G6PD(−) state is a selective disadvantage for hematopoietic stem cells; interestingly, this disadvantage is cell lineage–specific because it does not affect, for instance, fibroblasts. Finally, because selection might take place at other X-linked loci, heterozygotes exhibiting extreme phenotypes by analysis of G6PD may arise from selection acting on an allele at another locus (a “hitchhiking effect”), as has been suggested in a family with G6PD Ilesha. Evidence indicates that skewing may increase with age.
At the genomic level, the Gd gene ( Fig. 18-4, A ) consists of 13 exons, the first of which is noncoding. The total length of the gene is about 18.5 kilobases (kb), much of which (about 12 kb) consists of intron 2. The significance of this large intron is unknown; it may be important for efficient transcription or for processing because it is still the largest intron, even in the compressed version of the G6PD gene found in the puffer fish Fugu rubripes . The promoter region has a high proportion of guanine (G) and cytosine (C) residues (i.e., it is GC-rich), as found characteristically in other housekeeping genes. Deletion analysis has revealed that the “essential” portion of the promoter is only about 150 base pairs long. Within this region, two Sp1 binding sites have been identified, either of which is essential for promoter activity. This region and other regions are highly conserved between human and mouse, thus supporting the notion that they are important for gene regulation.
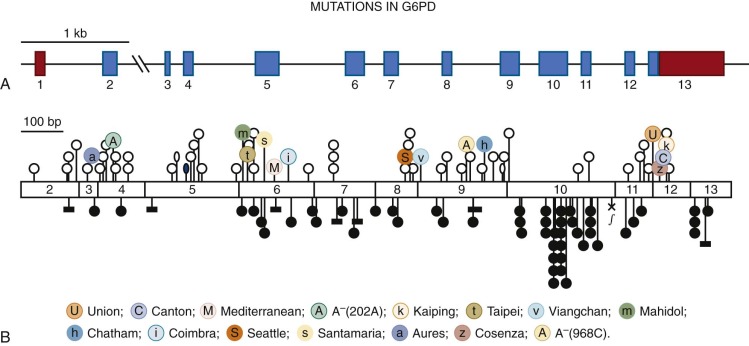
Features of G6PD in Red Blood Cells
Only one structural gene exists for G6PD, although a related autosomal hexose dehydrogenase also exists. Biochemical evidence is in keeping with the notion that the G6PD protein in red blood cells is the same as that in other somatic cells; thus when red blood cells are severely deficient in G6PD, this deficiency is also found to a greater or lesser degree in other somatic cells. However, a significant difference in the metabolism of G6PD arises from the characteristic inability of mature red blood cells to synthesize protein. As a result, whereas in most somatic cells G6PD is subject to turnover, in red blood cells any G6PD molecule undergoing denaturation or proteolytic breakdown cannot be replaced (this condition is true, of course, not only for G6PD but also for most other red blood cell enzymes ). In normal red blood cells, the decay of G6PD approximates an exponential (see Fig. 18-6 ) with a half-life of about 60 days, although it has been claimed that it may more closely approximate a two-slope curve with very fast breakdown when reticulocytes mature to erythrocytes and much slower breakdown subsequently. The age dependence of red blood cell G6PD activity is so characteristic that it can be almost regarded as a marker of red blood cell age. In normal blood, reticulocytes have about five times more activity than the 10% oldest red blood cells.
Molecular Basis of G6PD Deficiency
In principle, genetically determined deficiency of G6PD, like that of any other protein, might be attributed either to quantitative changes, such as mutations that affect the amount of the enzyme but not its structure, or to qualitative changes, such as mutations that affect the structure of the enzyme and hence its stability or catalytic efficiency. Extensive investigations of G6PD from G6PD-deficient cells, mostly carried out before the sequence of G6PD was known, revealed that (1) enzyme activity, even when severely reduced (sometimes to less than 1% of normal), is never completely absent and (2) enzymic properties ( K m , K i , activity on substrate analogues, and thermostability, for example) are often different from those of the normal enzyme (i.e., G6PD deficiency is associated with qualitative abnormalities). These data are consistent with point mutations in the coding region. In fact, from a database of 186 mutant alleles that are now known, a reasonably clear pattern has emerged (see Table 18-2 ).
- 1.
In nearly all G6PD variants there is a single amino acid replacement caused by a single missense point mutation.
- 2.
In several of the cases in which two amino acid replacements are found, one of them is that of G6PD A (N126D). Because this nondeficient G6PD variant is polymorphic in Africa, the most likely explanation for the presence of two mutations in cis is that a second point mutation has taken place in a Gd A gene.
- 3.
Ten internal gene deletions have been discovered, all of them in frame (see Table 18-2 ); we can presume that deletions causing frameshifts would be lethal.
- 4.
Only two mutations affecting splicing have been discovered thus far.
- 5.
The majority of the mutations (see Fig. 18-4, B ) in the database are sporadic, and most of them have been detected because they result in G6PD deficiency and CNSHA by causing sufficient loss of activity in red blood cells to become limiting for their in vivo survival. Sporadic variants associated with CNSHA are not likely to spread. However, in several instances the same variant has been encountered recurrently (for instance, we have found G6PD Tokyo in Scotland, G6PD Guadalajara has turned up in Japan and Belfast, and G6PD Nara has been observed at least three times). That the same mutation may be found recurrently in people who are almost certainly not ancestrally related is not trivial; these observations corroborate the notion that specific subtle constraints must exist whereby a particular G6PD variant has a distinctly severe clinical expression but remains compatible with life.
- 6.
The majority of known polymorphic mutations ( Fig. 18-5 ) again are associated with G6PD deficiency, and overwhelming evidence indicates that they have become polymorphic as a result of malaria selection (discussed later in this chapter). For these mutations we can visualize more stringent constraints; indeed, although still causing deficiency in red blood cells, they must not affect them so severely that the advantage with respect to malaria is outweighed.
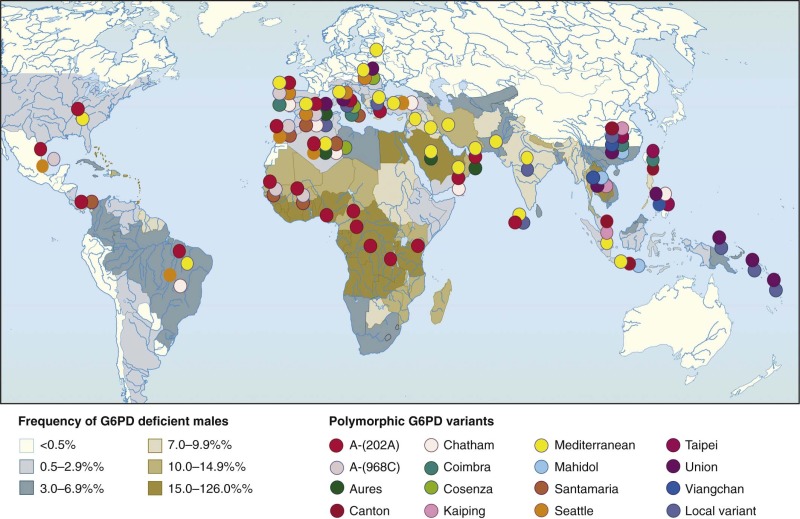
Figure 18-5
A world map of glucose-6-phosphate dehydrogenase (G6PD) deficiency. The shadings indicate the overall prevalence of G6PD deficiency in individual countries; the distribution of individual G6PD variants is shown by colored symbols.
(Modified from WHO Working Group: Glucose-6-phosphate dehydrogenase deficiency. Bull World Health Organ 67:601–611, 1989; and Tan KL, Boey KW: Clinical experience with phototherapy. Ann Acad Med Singapore 18:43–48, 1989.)
- 7.
The pattern of G6PD mutations, whereby null mutations (such as nonsense mutations, frameshifts, or large deletions) are conspicuously absent, is in striking contrast to that seen in other inherited disorders, such as the thalassemias, hemophilia, and muscular dystrophy. An important functional difference between these conditions and G6PD deficiency is that the former result from mutations in tissue-specific genes, whereas Gd is a housekeeping gene. When a tissue-specific gene is totally inactivated by a mutation, it may cause severe disease but will not necessarily have interfered with embryonic development. By contrast, we know that G6PD-null mutations are lethal to the mouse embryo ; they probably occur in humans, but we never see them because they must be lethal in the human embryo as well. Some clue regarding what mutation can cause G6PD deficiency has come from alignment of the amino acid sequence of G6PD from 42 different organisms. A striking correlation was found between the amino acid replacements that cause G6PD deficiency in humans and the sequence conservation of G6PD. Interestingly, two thirds of such replacements are in highly and moderately conserved (50% to 99%) amino acids, and relatively few are in fully conserved amino acids (where they might be lethal) or in poorly conserved amino acids, where presumably they simply would not cause G6PD deficiency.
- 8.
The biochemical mechanism of G6PD deficiency is strongly contingent on the fact that in normal red blood cells, G6PD decreases exponentially as red blood cells age, with a half-life of about 50 days (as discussed earlier in this chapter). Because G6PD is an oligomeric globular protein, it is not surprising that many point mutations producing replacement of individual amino acids can further decrease the stability of G6PD; in fact, for most mutants, enzyme instability is the main mechanism of enzyme deficiency ( Fig. 18-6 ). In a few cases (e.g., G6PD Orissa and G6PD Mahidol ), altered catalytic function (instead of or in addition to protein instability) may be the main mechanism of G6PD deficiency. The instability is most extreme when the binding of structural NADP is compromised or when the amino acid involved is at the interface between the two subunits of G6PD; in such cases the dimer does not form or falls apart. These situation apply to G6PD mutants that cause the most severe clinical phenotypes (that of CNSHA, as will be discussed later).
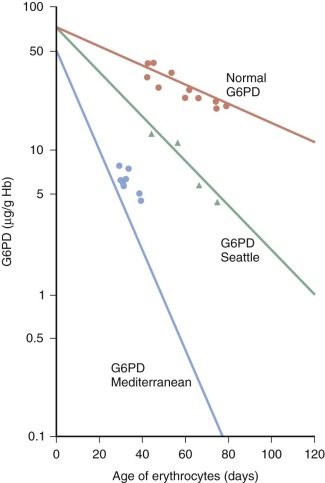
Figure 18-6
The main mechanism of glucose-6-phosphate dehydrogenase (G6PD) deficiency in red blood cells is in vivo instability of mutant enzyme. For many G6PD variants, such as the two studied here, this is essentially acceleration of a process that takes place normally with the aging of red blood cells in the circulation. Hb, Hemoglobin.
(Modified from Morelli A, Benatti U, Gaetani GF, et al.: Biochemical mechanisms of glucose-6-phosphate dehydrogenase deficiency. Proc Natl Acad Sci U S A 75:1979–1983, 1978.)
| Type of Mutation | Type of Mutation | No. of Mutants | Comments |
|---|---|---|---|
| Missense point mutations | Single | 159 | At least 35 of these mutations are polymorphic |
| Two | 13 | 6 of these mutations share the N126D mutation found on its own in the polymorphic nondeficient G6PD A: all of these are polymorphic, and 3 of them are the so-called G6PD A− common in Africa and in people of African descent 1 has the G6PD Canton mutation plus another mutation 1 of these has two mutations, one of which alone is known to cause CNSHA, and the other may aggravate the clinical picture | |
| Three | 2 | One of these has two mutations characteristic of G6PD A− plus a third mutation causing CNSHA | |
| In-frame deletions | 1 amino acid | 7 | None of these are polymorphic; all are associated with CNSHA |
| 2 amino acids | 1 | ||
| 6-8 amino acids | 2 | ||
| Affecting splicing | 2 | None of these are polymorphic; all are associated with CNSHA |
Recent studies on the rare disease incontinentia pigmenti have revealed the remarkable fact that the underlying gene, called IKBKG/NEMO , overlaps with Gd, is transcribed in the opposite direction, and shares one of its two promoters with Gd. Some patients with incontinentia pigmenti have large deletions that remove four exons of G6PD ; however, no G6PD deficiency has been reported in this unique situation because all affected patients are heterozygotes and presumably in the blood there is overwhelming selection in favor of the cells in which the active X chromosome has the normal Gd allele.
Epidemiology of G6PD Deficiency
The geographic distribution of G6PD deficiency is rather extraordinary in two ways. First, it has a very high prevalence overall, with more than 500 million people estimated to be involved (most of them mostly asymptomatic). Second, it has a high prevalence in many populations in most tropical and subtropical parts of the world because of malaria selection (see Fig. 18-5 and the last section in this chapter). Thus G6PD is prevalent in all five continents; however, it has never been reported in Amerindians, perhaps because they have been exposed to malaria only during the past three centuries. Very recently a new map of G6PD deficiency in malaria-endemic areas has been produced.
Clinical Manifestations of G6PD Deficiency
The most classic manifestation of G6PD deficiency is acute hemolytic anemia (AHA); in infants, however, another syndrome of great clinical and public health importance is neonatal jaundice (NNJ). CNSHA is a much rarer manifestation of G6PD deficiency and is the result of a lifelong hemolytic process. These different clinical manifestations are discussed in turn.
Acute Hemolytic Anemia
Clinical Picture
A child with G6PD deficiency is clinically and hematologically normal most of the time, and this status can be designated as a steady-state condition. However, a rather dramatic clinical picture can develop upon ingestion of fava beans (favism), during the course of infection, or after exposure to certain oxidative agents (see Table 18-3 ).
| Category of Drug | Definite Hemolysis | Possible Hemolysis |
|---|---|---|
| Antimalarial | Dapsone Primaquine Methylene blue † | Chloroquine Quinine |
| Analgesic | Phenazopyridine | Aspirin (high doses) |
| Antibacterial | Sulfamethoxazole/co-trimoxazole ‡ Sulfadiazine Ciprofloxacin Moxifloxacin Norfloxacin Ofloxacin Nalidixic Acid Nitrofurans: | Sulfasalazine |
| Nitrofurantoin | Chloramphenicol Isoniazid | |
| Other | Rasburicase Toluidine blue | Ascorbic acid Glibenclamide Vitamin K |
* The literature on this subject was critically reviewed by Youngster et al. in 2010, and we took this review into account when revising the table that appeared in previous editions. However, we believe that the notion of levels of evidence used to assess the efficacy of drugs cannot be applied in the same way to assess the safety of drugs. Therefore we believe that even individual case reports, if convincingly documented, must be taken seriously, and thus this table has some differences with respect to the article by Youngster et al.
† There has been a revival in the use of methylene blue (MB), together with artemisinin-based combinations, in the treatment of Plasmodium falciparum malaria, because MB also has a gametocytocidal action. At the doses used, MB has caused significant hemolysis in children in Africa ; most cases were not very severe.
‡ Atovaquone has been found to be a safe alternative in a glucose-6-phosphate dehydrogenase–deficient patient with Pneumocystis jiroveci infection ; this might apply to other protozoal infections.
Favism.
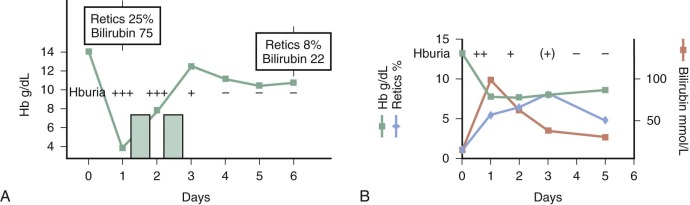
After a lag of hours since eating fava beans, the child may become fractious and irritable or subdued and even lethargic. Within 24 to 48 hours the child’s temperature is often moderately elevated. Nausea, abdominal pain, diarrhea, and rarely vomiting may be present. In striking contrast to these relatively nonspecific symptoms, the patient or a parent will observe, within 6 to 24 hours, the telltale and rather frightening sign of discolored urine ( Fig. 18-7 ). The urine will be reported as dark; as red, brown, or black; or as “passing blood instead of water.” Depending on experience, culture, and socioeconomic background, it will be said to resemble Coca-Cola, strong tea, or port wine. At about the same time, jaundice will become obvious. Physical examination may reveal little more than the signs corresponding to these symptoms. The child will be pale and tachycardic, and in severe cases evidence may be seen of hypovolemic shock or, less likely, heart failure. The spleen is usually moderately enlarged, and the liver may also be enlarged; either or both may be tender.
Drugs.
G6PD deficiency was first discovered by investigating a possible genetic basis for sensitivity to primaquine, and early studies have documented the clinical manifestation of primaquine-induced AHA in G6PD-deficient volunteers who were adults. In children with G6PD deficiency the largest reported series of AHA is not with primaquine but with dapsone. The children were from different parts of Africa, but because they all received the same dose, quantitative data could be obtained for the first time. In 119 G6PD-deficient (hemizygous or homozygous) children with malaria who received 7.5 mg/kg of dapsone over 3 days, the drop in hemoglobin averaged about 2 g/dL, with a nadir on day 7. An increase in neutrophils and a brisk reticulocyte response were noted, and the bilirubin peaked on day 3.
Laboratory Findings
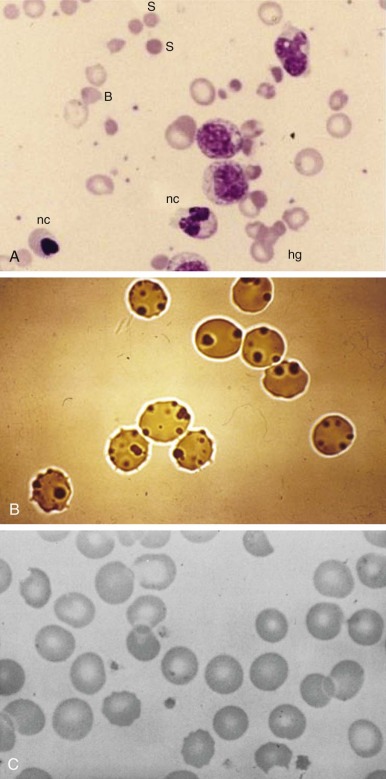
Anemia may range from moderate to extremely severe (hemoglobin values of 2.5 g/dL have been recorded). In the absence of other preexisting hematologic abnormalities, the anemia is normocytic and normochromic. The structure of the red blood cells may be striking ( Fig. 18-8, A ). Marked anisocytosis is often noted (reflected as a wide red blood cell size distribution on the electronic counter) because of the coexistence of large polychromatic cells and “contracted” cells, some of which can be frankly classified as spherocytes. Marked poikilocytosis is also noted with the presence of distorted red blood cells, “irregularly contracted” red blood cells, and red blood cells with apparently uneven distribution of the hemoglobin inside them (hemighosts). Although some of these appearances are probably smearing artifacts, electron micrographic evidence suggests that in some of the cells, opposing surfaces of the membrane have become “cross-linked.” Probably the most characteristic poikilocytes are those in which the cell margin literally appears dented, as though a portion has been plucked out or bitten away (“bite cells”; see Fig. 18-8, A ). The reticulocyte count is increased and may reach peaks of 30% or greater, with an elevated percentage of the immature reticulocyte fraction, reflecting a prompt and effective bone marrow response. Careful inspection of reticulocyte preparations may reveal inclusion bodies different from those normally seen in reticulocytes; these bodies are discrete, round, and 1 to 3 µm in diameter, and they usually appear to be leaning, from the interior, against the cell membrane. These inclusions are more clearly displayed by supravital staining with methyl violet and are referred to as Heinz bodies (see Fig. 18-8, B ). They consist of precipitates of denatured hemoglobin and are the vivid manifestation of the oxidative insult that this protein and the cell itself has experienced. However, Heinz bodies are a very transient finding because they tend to be promptly “pinched off” by the spleen (thus giving rise to bite cells), and the red blood cells that contain them are very rapidly removed from the circulation. Haptoglobin is reduced to the point of being undetectable. In severe cases it is possible to demonstrate free hemoglobin in plasma ( hemoglobinemia ). The white blood cell count is usually moderately elevated, with a predominance of granulocytes. The platelet count may be normal, increased, or moderately decreased. The unconjugated bilirubin level is elevated, but “liver enzyme” levels are generally normal. Test results of the dark urine are strongly positive for blood because of the presence of free hemoglobin (see the differential diagnosis of this sign in Table 18-4 ).
| Condition | Circumstances | Diagnostic Approach |
|---|---|---|
|
|
|
Clinical Course
In the majority of cases the hemolytic attack, even if severe, is self-limited and tends to resolve spontaneously (see Fig. 18-7, B ). Depending on the proportion of red blood cells that have been destroyed, the anemia will be more or less severe, but the hemoglobin level may be back to normal in 3 to 6 weeks. However, in some cases the decrease in hemoglobin is so precipitous that blood transfusion is required to save the child’s life. The proportion of such cases will depend primarily on (1) the dose of the offending agent, (2) concomitant pathology, and (3) the previous hemoglobin level. Although the blood urea level may be transiently elevated, the development of renal failure in children is exceedingly rare, even in the presence of massive hemoglobinuria (see Fig. 18-7, A ).
Diagnosis
With a history of fava bean ingestion or of administration of a potentially hemolytic drug (see Table 18-3 ) followed by hemoglobinuria (see Table 18-4 ), the diagnosis is almost always straightforward, and it can be made quite confidently even before obtaining the final proof that the patient is deficient in G6PD (discussed later in this chapter). If the hemoglobinuria has already subsided and the history is uncertain, one is faced instead with the much wider differential diagnosis of AHA. Negative findings of a direct antiglobulin test will militate against autoimmune hemolytic anemia. In endemic areas it will be important to exclude malaria or the much rarer babesiosis. In persons with hemolytic-uremic syndrome, the red blood cell structure is different, and evidence of impaired renal function will be seen. In all cases, demonstration of G6PD deficiency will be conclusive, and in uncertain cases it will be crucial.
Pathophysiology
The clinical picture of AHA in a G6PD-deficient child is a vivid expression of the fact that, as we know already, hemolysis results from the action of an exogenous factor on latently but intrinsically abnormal red blood cells. Hemoglobinemia and hemoglobinuria unambiguously indicate that the hemolysis is at least in part intravascular. At first approximation we can visualize the following sequence of events: (1) an oxidative agent causes conversion of GSH to oxidized glutathione; (2) because of the limited capacity of G6PD-deficient red blood cells to regenerate GSH, their GSH reserve is rapidly depleted; (3) once GSH is exhausted, the sulfhydryl groups of hemoglobin and probably other proteins are oxidized to disulfides or sulfoxides; and (4) coarse precipitates of denatured hemoglobin cause irreversible damage to the membrane, and the red blood cells lyse or become prey to macrophages.
Not all of these steps from oxidative attack to final hemolysis have been fully documented in vivo, in part because of one major difficulty: in the course of AHA the red blood cells sampled from the patient are obviously, at any given stage, those that have not yet hemolyzed . However, GSH depletion is a most characteristic finding when red blood cells are subjected to oxidative challenge, and in one careful study it has been demonstrated that during an attack of favism, the first measurable biochemical change is a decrease in NADPH, followed by a decrease in GSH, in keeping with stages 1 and 2 described earlier. Heinz bodies (see Fig. 18-8, B ) are the visible expression of stage 3. Stage 4 is less clearly defined, although studies suggest that the abnormal proteolytic activity is associated with increased levels of intracellular calcium, as well as binding of hemichromes (arising from denaturation of hemoglobin) to band 3 molecules.
Even though intravascular hemolysis in persons with AHA that is associated with G6PD deficiency is important in pathophysiology and diagnosis, a substantial proportion of the hemolysis is extravascular (as shown by the enlarged spleen). Probably the most severely damaged red blood cells hemolyze in the bloodstream on their own, whereas less severely damaged red blood cells will be recognized as abnormal by macrophages and will undergo extravascular hemolysis in the reticuloendothelial system. This process has been referred to as an example of an “innocent bystander” phenomenon (although the red blood cells, by virtue of being deficient in G6PD, are not that innocent), in which complement and immunoglobulins may be involved. Finally, it is important to remember that the red blood cell destruction in AHA associated with G6PD deficiency is an orderly function of red blood cell age (see Fig. 18-6 ). The oldest red blood cells with the least G6PD are the first to hemolyze, and the hemolytic process progresses upstream toward cells with more and more G6PD. As a result there is a selective enrichment in red blood cells that, despite being genetically G6PD deficient, have relatively higher levels of G6PD. This phenomenon can be so marked with certain G6PD variants that patients in the posthemolytic state are found to be relatively resistant to further challenge; thus the patient may be in a state of compensated hemolysis.
Triggers and Mechanism of Hemolysis
After primaquine, numerous other drugs have been reported as being potentially dangerous in G6PD-deficient persons ( Table 18-3 ). There is no obvious similarity in chemical structure among all of these substances, but they have in common the ability to stimulate the pentose phosphate pathway in red blood cells, which must mean that they are able to oxidize NADPH directly or indirectly. The components of fava beans responsible for hemolysis have been identified as vicine and convicine, two β-glycosides that generate the aglycones divicine and isouramil. These compounds, in the course of their auto-oxidation, produce free radicals, which in turn oxidize GSH and thereby activate the chain reaction of events previously outlined. The drugs listed in Table 18-3 , or their metabolites, act in a similar way. Henna dyes, which are used as cosmetics in several populations, mainly in Asia, can also cause serious hemolysis after absorption through the skin or ingestion (apparently to induce an abortion ).
An intriguing feature of AHA associated with G6PD deficiency is its erratic character, which is more conspicuous with certain agents than with others. For instance, it is estimated that in adults, ingestion of fava beans does not trigger AHA in more than 25% of cases, and even in the same person, favism may occur on one occasion but not on another. One obvious factor must be the amount of fava beans ingested (in relation to body mass). Another is the quality, with raw fava beans more likely to cause favism than cooked, frozen, or canned fava beans. Perhaps even more important is the finding that the glycoside content is a function of the maturity of the beans, with young, small beans being much richer (as well as more tasty!). With respect to drugs, primaquine causes hemolysis regularly, but aspirin does so only sometimes ; once again, the dosage must be important, but additional genetic or acquired factors affecting the metabolism of the drug may play a role. With regard to infection, the best documented precipitants are hepatitis, pneumonia, typhoid fever, and recently brucellosis and Clostridium difficile (in newborns ); however, viral infections of the upper respiratory or gastrointestinal tract may also cause hemolysis. Apparently trauma also can be a trigger. It has been reported that in a group of patients who had been involved in major road traffic accidents, the frequency and severity of infection were significantly higher in patients who were G6PD deficient, and more severe anemia also occurred in this group.
Release of peroxides during phagocytosis of bacteria by granulocytes might explain the hemolytic process in persons with bacterial infections. In this respect it is possible that hemolysis has sometimes been attributed to drugs used for treating infection when it should have been blamed on the infection itself.
Treatment
AHA in a child may be a diagnostic problem that, once solved, does not require any specific treatment at all; in other cases, the child’s condition may be considered a medical emergency requiring immediate action. The most urgent question is whether a blood transfusion is needed. It is difficult to give absolute directives, but the following guidelines may be useful :
- 1.
If the hemoglobin level is less than 7 g/dL, the child should undergo a transfusion immediately.
- 2.
If the hemoglobin level is less than 9 g/dL and evidence indicates persistent brisk hemolysis (hemoglobinuria), immediate blood transfusion is also indicated.
- 3.
If the hemoglobin level is greater than 9 g/dL but hemoglobinuria persists or if the hemoglobin level is between 7 and 9 g/dL but the child has no hemoglobinuria, the child should be kept under close observation for at least 48 hours and should undergo a transfusion if either condition 1 or 2 develops.
The most important complication that may require treatment is acute renal failure, which is exceedingly rare in children.
Neonatal Jaundice
From an epidemiologic point of view, it is noteworthy that the frequency of NNJ varies widely in different populations; in many populations in which G6PD deficiency is prevalent, the rate of pregnancies at risk for Rhesus incompatibility happens to be low. In addition, because Rhesus-related hemolytic disease of the newborn (HDN) is disappearing thanks to the implementation of appropriate prophylaxis, one can expect G6PD-related NNJ to be generally on the increase, at least in relative terms. G6PD deficiency has been well recognized as a major cause of NNJ in countries as disparate as Greece, Nigeria, and Taiwan. During the past decade in the United States it has been increasingly recognized that G6PD deficiency is an important etiologic factor of severe NNJ, including kernicterus.
Clinical Features
NNJ related to G6PD deficiency is very rarely present at birth; it has a peak incidence between day 2 and day 3. Jaundice is present more often than anemia, and the anemia is very rarely severe. For this reason the terms HDN and NNJ cannot be regarded as interchangeable, at least in the context of G6PD deficiency. The severity of G6PD-related NNJ varies from being subclinical to imposing the threat of kernicterus if not treated, and thus prompt recognition of the problem is extremely important to avoid crippling neurologic sequelae.
Nature of the Association between G6PD Deficiency and Neonatal Jaundice
It is not fully explained why NNJ develops in some but not all G6PD-deficient newborns. Several clinical studies have established beyond any possible doubt that the association is statistically much higher than could be expected by chance ( Table 18-5 ). However, because not all G6PD-deficient newborns have NNJ ( Table 18-6 ), it is likely that genetic or environmental factors (or both) are involved in addition to G6PD deficiency and that the same factors that are more extreme can also make the NNJ more severe. NNJ is not the prerogative of some G6PD variant because it is prevalent in widely remote parts of the world (e.g., Nigeria, Sardinia, Singapore, and China ) in which the Gd alleles underlying G6PD deficiency are different; moreover, within both Sardinia and Taiwan, NNJ has been found in babies with any of several different G6PD variants. The possibility that NNJ correlates with the quantitative level of residual G6PD activity is controversial. Specific features of erythrocytes in newborns such as elevated levels of ascorbic acid, depressed activity of glutathione reductase, and low levels of vitamin E, GSHPX, and other enzymes could contribute to the degree of jaundice. A dissociation has been reported between hyperbilirubinemia and anemia in G6PD-deficient neonates. Indeed, in one series there was no difference in the distribution of hematocrit values in cord blood and on day 3 between G6PD-deficient newborns with and without jaundice, thus suggesting that to a large extent this jaundice may be of hepatic origin rather than hemolytic. In keeping with this notion, measurements of the bilirubin production-conjugation index have demonstrated that the NNJ in otherwise healthy G6PD-deficient neonates depends more on inefficient bilirubin conjugation than on hemolysis. The UGT1A mutation characteristic of Gilbert syndrome is associated with a much higher risk for NNJ and kernicterus in G6PD-deficient babies. In addition, environmental factors can certainly exacerbate NNJ, in some cases by causing hemolysis in individual G6PD-deficient newborns; such factors include prematurity, breastfeeding, inhalation of naphthalene (camphor balls), acidosis, hypoxia, an infection such as viral hepatitis, use of oxidant drugs, and ingestion of drugs or fava beans (favism in utero ) by the mother before delivery.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


