Syndromic
Nonsyndromic
MEN1
FIHPT
MEN2
FHH
MEN4
NSHPT
HPT-JT
ADMH
Table 14.2
Features of primary hyperparathyroidism in hereditary syndromes
Penetrance in adult gene carriers | Age onset | Pathology of parathyroid glands | Parathyroid cancer | Associated tumors | Mutaded genes | |
|---|---|---|---|---|---|---|
MEN1 | 90% | 25–30 | Multiple adenomas | Very Rare | Multiple | MEN 1 |
MEN2A | ~ 30% | 34 | Multiple adenomas | Very Rare | Multiple | RET |
MEN4 | ~ 80% | ~ 50 | Multiple adenomas | ? | Multiple | CDKN |
FHH | 100% | At birth | Normal or mild hyperplasia | None | None | CaSR |
NSHPT | 100% | At birth | Hyperplasia | None | None | CaSR |
HPT-JT | 80% | 32 | Single or multiple cystic tumors | 15 % | Multiple | HRPT2 |
FIPHT | ? | ? | Single or multiple tumors | ? | ? | Unknown |
Conversely from sporadic pHPT, in which the removal of all hyperfunctioning tissue (tumoral or hyperplastic) definitively cures the hypercalcemia and the persistence or recurrence of the disease is very rare, definitive surgical cure of the familial forms is theoretically possible only if all the parathyroid tissue (either hyperfunctioning or normal) is excised, because all the parathyroid cells harbor the genetic mutation; therefore they are at risk to maintain or cause the recurrence of pHPT. However, the involvement of the parathyroid glands can be gradual and progressive, meaning that most of the time the HPT can be resolved by a limited or subtotal parathyroidectomy, as the main objective of surgery is to reach normocalcemia for as long as possible, avoiding permanent hypoparathyroidism [4]. A bilateral neck exploration is usually adopted to research all the parathyroid glands and to assist the decision on the most appropriate treatment. Preoperative assessment for determining the localization of the pathological parathyroid glands and selecting the type of surgery is probably less important than in the case of sporadic HPT. Even if all the imaging studies are utilized, only 68% of the enlarged parathyroid glands can be detected [5]. However, considering the high frequency of supernumerary or ectopic parathyroid glands in these forms of HPT, preoperative noninvasive localizing exams can be useful in at least 7% of the patients [5]. The localizing exams must be limited to ultrasonography, able to detect most ectopic parathyroid glands outside the mediastinum, and 99mTc-sestamibi scan, which can identify the mediastinal or undescended parathyroid glands. A more sophisticated preoperative assessment (utilizing one or more of the following studies: CT scan employing a four-dimensional technique, MRI, PET scan, venous sampling for parathyroid hormone [PTH] dosage, arteriography, fine needle biopsy) becomes mandatory for the recurrence of hereditary pHPT, since in this event a focused approach reduces the risk of damage of the recurrent laryngeal nerves or the other cervical or mediastinal nervous or vascular structures [4].
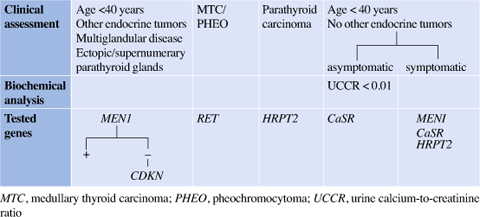
Table 14.3
Genetic screening of familial primary hypeparathiroidism based on clinical approach
14.2 MEN Syndromes
Multiple endocrine neoplasia (MEN) syndromes are rare disorders characterized by variable combinations of primary endocrine tumors in at least two different endocrine organs and the occurrence of other nonendocrine tumors or disorders. The MEN described until now are: type 1 (approximately one case in 30,000 births) usually presenting with various combinations of parathyroid adenoma, enteropancreatic NET and pituitary adenoma; type 2 (one case in 20,000 births) characterized mainly by medullary thyroid carcinoma (MTC) and pheochromocytoma; and type 4 (until now no more than ten patients are referred in the literature) showing a pathological spectrum similar to that of MEN1 (Table 14.2). MEN2 includes three clinical subtypes: MEN2A (approximately 90%); MEN2B, also defined as MEN3 (approximately 5%); and familial medullary thyroid carcinoma (FMTC). pHPT is found only in MEN2A [6].
The “multiple” definition refers both to the presence of multiple tumors in the involved endocrine tissue (the tumors can be synchronous or metachronous in the target organ) and to the development of tumors in different endocrine organs (e.g. parathyroid gland and pancreatic islet tumor in MEN1 syndrome or MTC and pheochromocytoma in MEN2A).
Two different forms of MEN, apparently sporadic and familial (more common), have been described. The apparently sporadic form is represented by a single patient (usually the “propositus”) with at least two of the most frequently observed MEN-related endocrine tumors. The familial form consists of a MEN patient with first-degree relatives either showing at least one of the characteristic endocrine tumors or harboring a mutation in the corresponding gene.
The penetrance of the various tumors is largely different: in MEN1 the most frequent tumors are the parathyroid adenomas followed by pancreatic NET, while in MEN2A the MTC is found in almost all subjects harboring the mutation of the RET gene, but parathyroid gland tumors are rare [6].
In contrast to nonhereditary endocrine tumors, MEN tumors usually arise at least 20–30 years in age earlier than sporadic tumors. The exception is represented by the age at onset of pituitary tumors, which is similar for sporadic and MEN1-associated pituitary tumors [7, 8]. The majority of the tumors have precursors represented by hyperplastic modifications of endocrine tissues, are benign at the moment of the diagnosis, and their malignant progression can be absent or may take several years. This long interval allows therapeutic action against the tumors, either by drugs controlling the endocrine symptoms and/or the neoplastic growth, or by surgery removing the target organ. It is important to know that, in the absence of therapeutical intervention, malignant pancreatic islet tumors and thymic carcinoid tumors are responsible for the death of MEN1 patients at a significantly younger age than that occurring in unaffected individuals [9].
14.2.1 MEN1 pHPT
MEN1 is an autosomal-dominant inherited syndrome due to germline mutations in the MEN1 gene. The MEN1 gene is a tumor suppressor gene located at chromosome 11. It consists of ten exons with a 1830-bp coding region that encodes the 610 amino acid protein called menin. MEN1 pHPT represents the most common familial cause of pHPT, accounting for approximately 2–4% of all cases of pHPT. More than 1000 different germline and somatic MEN1 gene mutations and 20 polymorphisms have been identified. The tumor susceptibility results from germline inactivation [6, 10].
The most common and the first endocrine manifestation of MEN1 syndrome is pHPT. Different from the sporadic counterpart, the age of occurrence is 20–30 years earlier (the most common age of onset is around 25 years) and its prevalence is similar in both sexes. The penetrance of HPT is very high, reaching 100% within 50 years of age [6]. Patients remain asymptomatic for a long period, but a progressive reduction of bone mass with an increased likelihood of skeletal fractures can be observed, especially in the affected women beginning from 35 years of age [11]. Hypercalciuria can cause nephrocalcinosis, renal stones, and progressive renal insufficiency. Hypercalcemia may increase the secretion of enteropancreatic hormones from NET, and particularly of the gastrin from duodenal gastrinomas, precipitating or exacerbating Zollinger-Ellison syndrome. The association of the MEN1 gastrinomas to parathyroid hyperfunction represents a more virulent form of pHPT [12].
It is recommended that mutant gene carriers undergo biochemical screening (serum calcium, PTH, chromogranin A, prolactin, IGF-1 and gastrointestinal hormones) at least once per year beginning in early childhood, since the disease can develop by the age of 5 years, and should continue for life, since the disease can develop very late in some individuals (in the seventh or eighth decade) [13].
Even if all the parathyroid glands are affected, different enlargement of the parathyroid glands is observed at the time of diagnosis. One, or more than one, of the parathyroid glands can have a normal aspect, normal volume, and sometimes also normal histological pattern [14–16] (Fig. 14.1). Sometimes the parathyroid glands have a characteristic polylobulated morphology different from the ovoid aspect of sporadic adenoma (Fig. 14.2). If the pHPT is not appropriately recognized as part of MEN1 syndrome and is confused with a sporadic single or double parathyroid gland adenoma, it is possible that inadequate treatment is given. A peculiar characteristic of this type of pHPT is the presence of supernumerary parathyroid glands (up to 30% of the affected patients) and the ectopic site of the parathyroid glands that is localized in the thymus, in the anterior mediastinum, or, more rarely, in the thyroid or the carotid sheath (Fig. 14.3). In particular, the most frequently observed site of ectopic parathyroid glands is that of the thymus, which is variable from 4.9% to 30% [17–19]. Also, the presence of parathyroid nests embedded within the fatty tissue surrounding the trachea, the esophagus, and the carotid artery have been described, explaining the potential growth of recurrent HPT also after a total parathyroidectomy [20]. Malignant progression is considered extremely rare. The prevalence of parathyroid carcinoma is 0.28% according to the revision of the database of 348 MEN1 patients followed at the Mayo Clinic [21]. However, in the last 20 years, 11 parathyroid carcinomas have been observed in patients affected by MEN1 syndrome with an age of expression around age 50, and with an equal distribution between the sexes. Parathyroid carcinoma in MEN1 is only rarely associated to severe hypercalcemia, and metastasis or local infiltration are found in a minority of cases [21]. In the majority of parathyroid carcinomas, the diagnosis is posed only on the basis of the histological features, well recognizing the limits that this assessment presents in most endocrine neoplasms.
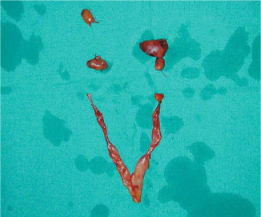
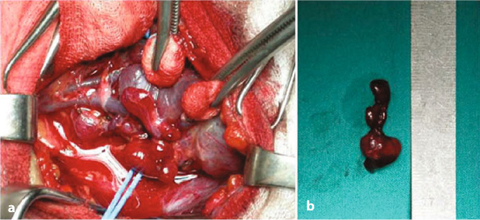
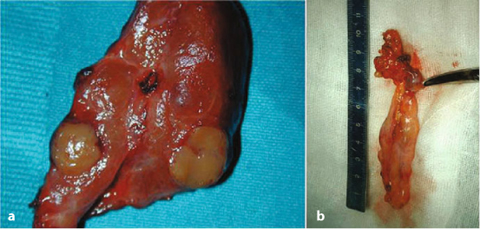
Fig. 14.1
Surgical specimen of a total parathyroidectomy and thymectomy performed for pHPT in MEN1 syndrome. The diverse size among the different parathyroid glands is evident
Fig. 14.2
MEN1 patients affected by pHPT. a Surgical view of the left superior parathyroid gland, which is compressed by the inferior thyroideal artery; b Image of the surgical specimen characterized by lobulated morphology
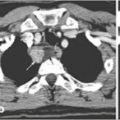
Fig. 14.3
a MEN1 patient with presence of an intrathyroideal parathyroid adenoma. b MEN1 patient with presence of a parathyroid adenoma in the thymic horn
14.2.1.1 Surgical Options
The preferred treatment is surgical removal of the parathyroid glands, but no general agreement has been reached about the appropriate timing of surgery and the number of parathyroid glands to excise. Severity of symptoms, alteration of biochemical exams (calcemia, calciuria, PTH), kidney stones, and decrease of bone mass density (BMD) can contribute to the decision regarding the time of surgery. Parathyroidectomy should adequately cure the hyperparathyroidism, avoiding its persistence or early recurrence, but in general avoid the hypoparathyroidism, which, it is well known, can be difficult to control and worse than a mild HPT. Several options have been proposed: parathyroidectomy limited to enlarged glands, subtotal parathyroidectomy (sPTx: 3, 3½ or 3 parathyroid gland removal), unilateral parathyroidectomy (both glands from the ipsilateral neck associated to the cervical thymic horn), or total parathyroidectomy (tPTx) with autograft of parathyroid gland fragments in the forearm muscle or subcutaneous tissue of the nondominant arm (Fig. 14.3). At the time of parathyroidectomy, the thymus should be removed, given the chance of finding an ectopic parathyroid gland and preventing the growth of a thymic carcinoid tumor, especially in males. However, cervical thymectomy cannot adequately remove the part of the thymus located deep in the mediastinum, and its prophylactic role for thymic carcinoid is doubtful [22–24]. There are several reports (all in men) of aggressive thymic carcinoid in patients submitted for thymectomy at the moment of the parathyroidectomy for MEN1 HPT. It is probable that residual mediastinal thymic tissue remains at a risk of developing carcinoid tumor. Removal of all the thymus is better performed by a mediastinal approach, either by a sternotomy or by thoracoscopy.
The choice between the many surgical options remains controversial. Observing the more consistent case list published in the literature, the removal of only the enlarged gland (usually 1 or 2) or a less than subtotal parathyroidectomy (<sPTx) exposes the patient to a high prevalence of persistence and recurrence of HPT. A meta-analysis of 12 studies comparing patients who had undergone <sPTx with patients who had undergone sPTx or tPTx showed a significantly higher risk of persistent or recurrent HPT after <sPTx [25]. However, the genotype of patients submitted to <sPTx can influence the surgical result. A retrospective study of the Dutch MEN1 database showed that patients with nonsense or frameshift mutations in exons 2, 9, and 10 had a significantly lower risk of permanent or recurrent HPT [26]. If these observations are confirmed by additional studies, the genotyping will be useful to individualize the extent of initial parathyroidectomy for patients with MEN1 [27].
An evaluation of experience with sPTx vs. tPTx by a meta-analysis (Tables 14.4 and 14.5) showed that sPTx patients have the same risk of recurrent HPT as patients with tPTx, but a lower risk of permanent hypoparathyroidism.
Table 14.4
Results after subtotal parathyroidectomy in MEN1 hyperparathyroidism
Author | Year | Period | Patients (n.) | Mean follow-up (years) | Persistent HPT (%) | Recurrent HPT (%) | Permanent hypoPT (%) |
|---|---|---|---|---|---|---|---|
Edis et al. [28] | 1979 | 1959–76 | 55 | 3.9 | 13 | 0 | 35 |
Prinz et al. [29] | 1981 | 1955–76 | 12 | 9.5 | 33 | 0 | 25 |
Van Heerden et al. [30] | 1983 | 1960–83 | 45 | N.A. | 6.6 | 6.6 | 13 |
Goretzki etal. [31] | 1991 | 1986–90 | 18 | N.A. | 11 | 0 | 0 |
Hellman et al. [32] | 1992 | 1982–91 | 11 | 11.9 | 0 | 27.3 | 27.3 |
Kraimps etal. [17] | 1992 | 1966–88 | 14 | 8 | 14 | 36 | 10 |
O’Riordain etal. [15] | 1993 | 1970–91 | 54 | 10 | 0 | 16.4* | 8 |
Janson et al. [33] | 1994 | 1971–92 | 4 | 9.9 | 0 | 25 | 0 |
Thompson et al. [34] | 1994 | 1972–92 | 14 | 20 | 7 | 7 | 0 |
Grant at al. [35] | 1994 | 1980–93 | 15 | 4.7 | 0 | 13.3 | 0 |
Nilsson et al. [36] | 1994 | 1971–92 | 2 | 9 | 0 | 0 | 0 |
Hellman et al. [37] | 1998 | 1969–96 | 9 | 7.3 | 22 | 44 | 0 |
Goudet et al. [9] | 2001 | 1986–97 | 73 | n.a. | n.a. | n.a. | n.a. |
Dotzenrath et al. [38] | 2001 | 1986–98 | 25 | 10 | n.a. | 8* | 12 |
Arnalsteen etal. [18] | 2002 | 1992–01 | 66 | 10 | n.a. | 33* | 12.7 |
Elaraj et al. [39] | 2003 | 1960–02 | 63 | 10 | n.a. | 51* | 26 |
Hubbard et al. [40] | 2006 | 1974–02 | 21 | 5 | 0 | 5 | 10 |
Norton etal. [12] | 2008 | 1970–05 | 41 | 7.9 | 12 | 44 | 10 |
Wadmann et al. [41] | 2010 | 1987–2009 | 11 | 9.8 | 0 | 18 | 45.5 |
Balsalobre et al. [42] | 2010 | 1980–2008 | 69 | 6.2 | 0 | 13 | 4.3 |
Heterman et al. [26] | 2012 | 1990–2009 | 23 | 4.3 | n.a. | 17 | 39 |
Nilubol et al. [43] | 2013 | 1997–2011 | 47 | n.a. | 7.7 | 20 | 11.5 |
Lairmore et al. [44] | 2014 | 1996–2012 | 17 | 7.5 | 6 | 24 | 12 |
Table 14.5
Results after total parathyroidectomy in MEN1 hyperparathyroidism
Author | Year | Period | Patients (n.) | Mean follow-up (years) | Persistent HPT (%) | Recurrent HPT (%) | Permanent hypoPT (%) |
|---|---|---|---|---|---|---|---|
Wells et al. [45] | 1980 | 1973–80 | 36 | 7 | 3 | 30 | 5.6 |
Malmaeus et al. [46] | 1986 | 1961–85 | 18 | 6.5 | 0 | 0 | 26 |
Hellman et al. [32] | 1992 | 1982–91 | 23 | 6.1 | 0 | 22 | 30 |
Janson et al. [33] | 1994 | 1971–92 | 6 | 9.9 | 0 | 0 | 0 |
Dralle et al. [47] | 1994 | 1976–92 | 4 | 6.3 | 0 | 0 | 50 |
Nilsson et al. [36] | 1994 | 1971–92 | 6 | 9 | 0 | 0 | 0 |
Hellmann et al. [37] | 1998 | 1969–96 | 15 | 10.2 | 0 | 20 | 47 |
Elaraj et al. [39] | 2003 | 1960–02 | 16 | 10 | n.a. | 16* | 46 |
Hubbard et al. [40] | 2006 | 1974–02 | 4 | 14 | 0 | 50 | 25 |
Norton et al. [12] | 2008 | 1970–05 | 9 | 9.9 | 0 | 55 | 22 |
Tonellietal. [16] | 2007 | 1990–06 | 45 | 6.5 | 0 | 11 | 22 |
Waldmann et al. [41] | 2010 | 1987–09 | 23 | 9.8 | 4 | 4 | 21.7 |
Pietermann et al. [26] | 2012 | 1990–09 | 32 | 4.3 | n.a. | 19 | 66 |
Montenegro et al. [19] | 2012 | 1987–11 | 75 | n.a. | 8 | 6.7 | 40 |
Lairmore et al. [44] | 2014 | 1996–12 | 15 | 7.5 | 0 | 13 | 7 |
sPTx is accompanied by a higher percentage of persistence and recurrence of HPT, but a lower percentage of transient or permanent hypoparathyroidism in comparison with tPTx and autograft of parathyroid tissue. The best results of tPTx and autograft are achieved when, during surgery, it is possible to assess the completeness of parathyroidectomy by measuring the value of PTH at the end of the procedure and, after that, implant fresh parathyroid tissue. Following this strategy the incidence of persistent HPT is greatly reduced, recurrence of HPT after 10 years is only 10%, but permanent hypoparathyroidism is observed in 22% of the patients [16].
Parathyroid autograft consists of putting 2–3 pieces (around 1 mm3) of the most normal parathyroid gland excised in pockets appointed either between the muscular fibers or in the subcutaneous tissue. There are no comparative studies determining which is the best site or the best number of fragments for auto-transplant. The most adopted method is to put 20-25 fragments in the forearm muscles [17, 44].
Recently, an attempt to give an answer to the most appropriate surgical treatment was evaluated in a randomized trial between subtotal and total parathyroidectomy [44]. Thirty-two patients were randomized from 1966 through 2012. At a mean follow-up of 7.5+5.7 years, the rate of recurrent HPT was 24% in patients treated with sPTx and 13% in patients treated with tPTx. The rate of permanent hypoparathyroidism was 12% in the sPTx group and 7% in the tPTx group. The occurrence of a second cervical operation is higher in sPTx patients (24% vs. 7%). These differences are nonsignificant because the power of this study was limited by the relatively limited number of the patients.
The recurrence rate after parathyroidectomy varies from 0% to 55%, and the time elapsed for recurrence is largely variable, from a few months to more than 10 years [17]. In order to limit or avoid the risk of permanent hypoparathyroidism, a less aggressive surgery has recently been proposed in young patients in whom preoperative localization studies (99mTc-sestamibi scan), ultrasonography and 4DTC) are concordant with the presence of 1-2 enlarged parathyroid glands on one side [48]. The surgery is performed through a small lateral cervical incision accessing the central neck and prevertebral plane lateral to the edge of the strap muscles with removal of both the ipsilateral parathyroid glands and the ipsilateral thymus. Ten patients have been treated with no recurrence and no hypoparathyroidism. However, the mean follow-up of these patients is only 19 months.
Risks for recurrence are: type of surgery (<sPTx is accompanied by a significantly higher rate of permanent and recurrent HPT); young age; and genotype (at least for patients operated on with <sPTx) [26, 44],
The rate of permanent hypoparathyroidism is related to the type of surgery. tPTx has a higher risk in comparison to sPTx. However, a consistent risk of hypoparathyroidism can be observed even in patients in whom <sPTx is performed. The rate is variable from 0 to 45. The cause of hypoparathyroidism is due to the failure of the parathyroid remnants left in situ that cannot function adequately because they are too small or not well vascularized. Cryopreservation of parathyroid tissue should be undertaken and eventually employed for autotransplantation. A higher risk of permanent hypoparathyroidism is observed if surgery is performed for recurrence, the rate of hypoparathyroidism increasing from 22 to 50% [16]. Even the time elapsed from surgery influences the rate of permanent hypoparathyroidism: the longer the follow-up, the lower the frequency of patients requiring calcium supplementation [19].
Recurrent HPT should be preferentially conducted by a focused parathy-roidectomy with limited exploration after accurate localization of the site of the residual parathyroid gland (or glands) in order to reduce the risk of damage to inferior laryngeal nerves in the scarred neck.
Considering the risk of a permanent hypoparathyroidism, it is suggested to cryopreserve some pieces of the parathyroid tissue for a successive implant. However, the cryopreserved tissue is less prone to remain viable and grow in comparison to fresh tissue.
14.2.1.2 Intraoperative PTH Monitoring
The rapid intraoperative PTH (ioPTH) assay has become an important diagnostic tool to guide parathyroid surgery. Its accuracy is reported in the order of 98% for the surgical treatment of sporadic adenoma when the Miami criterion is fulfilled [49]. It can be of value also in confirmation of the presence of a multiglandular HPT: the intraoperative PTH kinetic is usually different from that observed in single sporadic adenomas with a progressive decline of the circulating PTH values during the removal of the various hyperfunctioning parathyroid glands instead of a drastic drop. In recent years, experience with ioPTH has shown that differences in the methods and criterium used for indicating successful surgery have been adopted. Some authors prefer to do only a basal sample prior to skin incision, and one 10 minutes after removal of the last parathyroid gland [43], whereas other authors follow the PTH values several times during surgery until 1 hour after removal of the last parathyroid gland [16, 50]. The first method can cause false negative results due to a high increase of PTH during the surgical manipulation of the parathyroid glands. The second method is criticized for the cost. Failure of the ioPTH to identify patients who will develop persistent HPT is not rare because a predominant functioning gland is able to suppress the function of the other parathyroid glands, causing false positive results [51].
An important role of the ioPTH is to ascertain the removal of all functioning parathyroid tissue, allowing research of eventual supernumerary glands to be stopped, and implantation of fresh parathyroid fragments if tPTx + AT has been chosen [16, 18, 50]. The best predictive criterion for indicating a total parathyroidectomy is having a PTH value near the detectable limit, or also undetectable, 20 minutes after the removal of the parathyroid glands (6 pg/mL with the ICMA assay) [16]. Following this criterion, no false positive results have been observed in 48 MEN1 patients.
Related posts:
 Management of Primary Hyperparathyroidism
Management of Primary Hyperparathyroidism
 Clinical Manifestations of Primary Hyperparathyroidism
Clinical Manifestations of Primary Hyperparathyroidism
 Etiology and Pathogenesis of Primary Hyperparathyroidism and Hypercalcemias
Etiology and Pathogenesis of Primary Hyperparathyroidism and Hypercalcemias
 Surgical Anatomy of the Parathyroid Glands
Surgical Anatomy of the Parathyroid Glands
 Minimally Invasive Video-Assisted Parathyroidectomy
Minimally Invasive Video-Assisted Parathyroidectomy
 Preoperative Localization for Parathyroid Surgery in Primary and Secondary Hyperparathyroidism
Preoperative Localization for Parathyroid Surgery in Primary and Secondary Hyperparathyroidism
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


