Chapter Outline
PRINCIPLES OF CANCER PREDISPOSITION
IDENTIFICATION OF A CHILD WITH A CONDITION PREDISPOSING TO CANCER
Features of Family History Suggestive of a Heritable Predisposition to Cancer
Tumor Features Suggestive of a Heritable Predisposition to Cancer
Other Clinical Features Suggestive of a Heritable Predisposition to Cancer
Incidental Identification of a Child with a Condition Predisposing to Cancer
Genetic Testing for a Heritable Predisposition to Cancer
Management of Children with a Heritable Predisposition to Cancer
SPECIFIC SYNDROMES PREDISPOSING TO CANCER
WT1 -Related Wilms Tumor Predisposition Syndromes
Wilms Tumor, Aniridia, Genital Anomalies, Retardation Syndrome
Hereditary Paraganglioma/Pheochromocytoma Syndrome
Beckwith-Wiedemann Syndrome and Isolated Hemihyperplasia
Rhabdoid Tumor Predisposition Syndrome
DICER1 -Related Tumor Predisposition Syndrome
It is currently estimated that 1% to 10% of children with cancer develop the disease due to an underlying genetic predisposition. Over the last 3 decades, there has been an explosion in knowledge regarding the heritable causes of cancer, including the discovery of many genes that when mutated in the germline significantly increase the risk for tumor formation in children. This knowledge has allowed for a better understanding of the mechanisms leading to tumor formation and provided insights into the development of novel cancer therapies targeting defective genetic pathways. Through presymptomatic genetic testing, it is also now possible to identify children who are at increased risk for tumor formation and to initiate surveillance protocols with the intent to improve overall outcomes through the detection of early-stage tumors that are more readily cured with less invasive and/or toxic therapies. In this chapter, we review the principles of cancer predisposition, steps involved in the recognition and testing of children for heritable cancer syndromes, and concepts underlying tumor surveillance. We also describe the clinical and genetic features typifying specific syndromes and review current recommendations regarding management. As there are too many conditions to discuss in this chapter, we focus on several syndromes that are associated primarily with the development of solid tumors. For a review of additional childhood cancer syndromes, including those that increase the risk for hematologic malignant tumors, we refer the reader to several excellent recent reviews.
Principles of Cancer Predisposition
Seminal insights into cancer predisposition were gained in 1971 when Alfred Knudson developed the two-hit hypothesis to explain the epidemiology of retinoblastoma (RB), the most common pediatric eye tumor. Children with RB show two patterns of tumor formation, including: (1) early-onset disease characterized by bilateral eye involvement and autosomal dominant inheritance; or (2) later-onset disease typified by unilateral eye involvement with no family history of RB. According to the two-hit hypothesis, children with bilateral RB are at increased risk for tumor formation because they carry an altered copy of a growth regulatory gene within the cells of the body; this constitutional (germline) mutation is known as the first hit. Knudson proposed that if the second gene copy underwent an inactivating mutation within a developing retinal cell (i.e., the second hit), that cell would then become susceptible to tumor formation. Because every cell in a child with heritable RB carries the first hit, children with this form are likely to develop more than one tumor (multifocal and/or bilateral RB) at a very young age. In contrast, for those with nonheritable RB, both hits would need to occur within a single retinal cell. As these mutational events are exceedingly rare, it is expected that the process would take longer and be less likely to occur. Thus, children with nonheritable RB would be older at presentation and only develop unilateral tumors.
In 1986, Stephen Friend confirmed Knudson’s prediction by demonstrating that patients with bilateral RB harbor inactivating germline mutations in the RB susceptibility gene RB1 , the first tumor suppressor gene to be identified. Since this initial observation, many additional tumor suppressor genes have been cloned, and it is now clear that inactivating tumor suppressor gene mutations account for the majority of heritable cancers in humans. Other genetic mechanisms that contribute to heritable cancers include the presence of constitutional activating or “gain-of-function” mutations involving one copy of a growth-promoting oncogene, inactivating mutations in one or both alleles of an X-linked or autosomal recessive gene, and larger structural chromosomal abnormalities such as translocations, deletions, and duplications that disrupt or enhance the expression of a tumor suppressor gene or oncogene, respectively.
Identification of a Child with a Condition Predisposing to Cancer
Pediatric oncologists play an important role in recognizing hereditary cancer syndromes in children and their families. Toward this end, there are several possible clues that point to the existence of a condition that predisposes a child to cancer, including: (1) a positive family history of cancer; (2) the diagnosis of specific tumor types in the child; (3) tumors that involve paired organs; and (4) the presence of associated physical or syndromic features. Pediatric oncologists should consider each of these factors when evaluating a patient and refer the child to a geneticist or genetic counselor when there is a suspicion of an underlying cancer predisposition.
Features of Family History Suggestive of a Heritable Predisposition to Cancer
Features of the family history that are suggestive of a genetic predisposition to cancer include the presence in the child or his or her close relatives of one or more cancers presenting at a younger than expected age, involving both of paired organs (e.g., eyes, kidneys, or adrenal glands) or exhibiting multifocality (more than one discrete tumor within an organ). Other suspicious features include the existence of multiple individuals on the same side of the family (maternal or paternal lineage) with the same type of cancer or cancers known to cluster together in certain syndromes (e.g., the clustering of soft tissue and bone sarcomas, early-onset breast cancer, brain tumors, adrenocortical cancers, and leukemia in Li-Fraumeni syndrome [LFS]).
Pediatricians and pediatric oncologists should take a careful family history and generate a pedigree as a routine part of each patient’s initial clinic visit. Ideally, the pedigree should include information about the child being seen (the proband), as well as his or her first-degree relatives (parents and siblings), second-degree relatives (aunts, uncles, and grandparents), and third-degree relatives (first cousins). For any individuals who have developed a cancer, its type, site of origin, stage (laterality, focality), and the age at which it was diagnosed should be determined. It is important to note whether individuals have had more than one cancer, and if so, to distinguish whether multiple cancers represent recurrence of the initial tumor or development of a second primary malignant tumor. Finally, it is important to gather information about the ethnic background of the family, as some genetic syndromes manifest more commonly in individuals from specific geographic regions (as is the case with hereditary breast and ovarian cancer, which is more common in Ashkenazi Jewish individuals with ancestors from Eastern Europe). Information about the biologic relationship between parents can provide insights into the mode of inheritance. For example, if parents are closely related or both come from a similar geographic region, there is a greater chance for consanguinity and the presence of an autosomal recessive condition.
The importance of collecting a family history cannot be underestimated; however, this procedure can have its limitations. Some hereditary syndromes exhibit incomplete penetrance where not everyone with the inherited mutation will develop a cancer. Variable expressivity can be a complicating factor, with affected individuals manifesting in different ways within a given family. One must recognize that family histories evolve over time. A child may have an affected parent; however, this parent might be young or not yet have developed a cancer or other features typical of the condition. In some cases, affected family members may die of other causes before they develop a cancer. Finally, the family history may be unrevealing when there is a de novo germline mutation in an affected child, the presence of autosomal recessive or X-linked recessive conditions (where carriers are clinically unaffected), nonpaternity, or situations in which a child is adopted and there is little knowledge of the biologic parents’ family history. Due to these factors, it may be difficult to ascertain from a pedigree whether a specific syndrome is present. Nonetheless, the family history remains an important tool in recognizing conditions predisposing a child to cancer, and one should be obtained, regularly updated, and reviewed for the presence of a possible predisposition.
Tumor Features Suggestive of a Heritable Predisposition to Cancer
In addition to whether the tumor is unilateral or bilateral, unifocal or multifocal, specific tumor types should raise the suspicion of a syndrome predisposing to cancer, even in the absence of a positive family cancer history ( Table 42-1 ). RB is an excellent example as there is a hereditable component in 40% of patients. Additional tumor types associated with a high likelihood of an underlying genetic predisposition include: (1) Wilms tumor (WT), where features such as bilaterality, early age of onset, and genitourinary anomalies in boys indicate a higher likelihood of a germline WT1 mutation ; (2) pheochromocytoma (PCC) and paraganglioma (PGL), where more than 70% carry a germline mutation in SDHB or other associated PCC/PGL genes ; (3) atypical teratoid/rhabdoid tumor, where 35% carry a mutation in the INI1/SMARCB1 gene ; (4) pleuropulmonary blastoma (PPB), where more than 70% of cases in one study carried a germline DICER1 mutation ; (5) hepatoblastoma (HB), where up to 10% carry a mutation in the APC gene that is causative of familial adenomatous polyposis (FAP) ; (6) medullary thyroid cancer (MTC), where 20% to 25% carry a mutation in the RET protooncogene ; (7) medulloblastoma, where onset at an age younger than 5 years and desmoplastic histology are indicative of possible nevoid basal cell carcinoma syndrome (NBCCS, Gorlin syndrome) , where patients harbor germline mutations of PTCH1 ; and (8) adrenocortical carcinoma (ACC), choroid plexus carcinoma (CPC), and rhabdomyosarcoma presenting under the age of 3 years, where 50% to 80%, 35% to 100%, and 20% of children, respectively, carry germline mutations in the TP53 gene. The diagnosis of an adult-onset tumor in a child should also raise suspicion for a predisposing condition. For example, the presence of colorectal cancer could indicate a constitutional mismatch repair–deficiency (CMMR-D).
| Tumor Type | Predisposition Syndrome | Gene(s) |
|---|---|---|
| Adrenocortical carcinoma * | Li-Fraumeni syndrome (LFS) | TP53 |
| Astrocytoma | LFS | TP53 |
| Tuberous sclerosis | TSC1, TSC2 | |
| Atypical teratoid or rhabdoid tumor * | Rhabdoid tumor syndrome | SMARCB1/INI1 |
| Basal cell carcinoma | Nevoid basal cell carcinoma syndrome (NBCCS) | PTCH1 |
| Choroid plexus carcinoma * | LFS | TP53 |
| Cystic nephroma * Bilateral or multifocal | Pleuropulmonary blastoma (PPB) family tumor and dysplasia syndrome/ DICER1 syndrome | DICER1 |
| Endolymphatic sac tumors | von Hippel–Lindau syndrome (VHL) | VHL |
| Fibroma | Familial ademonatous polyposis (FAP) | APC |
| Gardner fibroma (desmoid fibroma) | ||
| Cardiac fibroma | NBCCS | PTCH1 |
| Hemangioblastoma | VHL | VHL |
| Glioblastoma | LFS | TP53 |
| Constitutional mismatch repair–deficiency syndrome (CMMR-D) | MLH1, MSH2, MSH6, PMS2 | |
| Hepatoblastoma | Beckwith-Wiedemann syndrome (BWS) | 11p15, CDKN1C |
| FAP | APC | |
| Leukemia | ||
| Pre-B acute lymphoblastic (low-hypodiploid) * leukemia | PAX5- associated familial leukemia | PAX5 |
| LFS | TP53 | |
| Other types † | CMMR-D | MLH1, MSH2, MSH6, PMS2 |
| Neurofibromatosis type 1 (NF1) | NF1 | |
| Lipoma | PTEN hamartoma tumor syndrome (PHTS) | PTEN |
| Malignant peripheral nerve sheath tumor | NF1 | NF1 |
| Medulloblastoma | FAP | APC |
| NBCCS | PTCH1 | |
| Neuroblastoma * Bilateral or multifocal | Hereditary neuroblastoma | ALK PHOX2B |
| Optic pathway glioma | NF1 | NF1 |
| Ovarian sex-cord stromal tumors | PPB family tumor and dysplasia syndrome/ DICER1 syndrome | DICER1 |
| Peutz-Jeghers syndrome (PJS) | STK11/LKB1 | |
| Papillary cystadenoma of epididymis or broad ligament | VHL | VHL |
| Paraganglioma (PGL)/pheochromocytoma (PCC) * | Hereditary PGL/PCC syndrome | SDHA, SDHB, SDHC, SDHD, SDHAF2, MAX, TMEM127 |
| Multiple endocrine neoplasia type 2 (MEN2) | RET | |
| NF1 | NF1 | |
| VHL | VHL | |
| Pineoblastoma | Hereditary retinoblastoma (RB) | RB1 |
| PPB * | PPB family tumor and dysplasia syndrome/ DICER1 syndrome | DICER1 |
| Retinoblastoma * | Hereditary RB | RB1 |
| Sarcoma | ||
| Liposarcoma | LFS | TP53 |
| Rhabdomyosarcoma | LFS | TP53 |
| PPB family tumor and dysplasia syndrome/ DICER1 syndrome | DICER1 | |
| Costello syndrome | HRAS | |
| Osteosarcoma | Hereditary RB | RB1 |
| LFS | TP53 | |
| Schwannoma | ||
| Acoustic neuroma * | Neurofibromatosis type 2 (NF2) | NF2 |
| Schwanommatosis | Rhabdoid tumor syndrome | SMARCB1/INI1 |
| Thyroid | ||
| Medullary * | MEN2 | RET |
| Nonmedullary | PHTS | PTEN |
| Wilms tumor | BWS | 11p15, CDKN1C |
| Bilateral or multifocal * Or with associated GU defects in males (hypospadias, undescended testicles) | WT1 -related syndromes | WT1 |
* Tumor types that warrant genetic testing even if there is no other personal or family history of cancer (where noted, only if tumor is bilateral or multifocal).
† Several other leukemia predisposition syndromes exist that are not discussed here.
Other Clinical Features Suggestive of a Heritable Predisposition to Cancer
Specific clinical features can provide clues to the diagnosis of an underlying cancer predisposition. These features include physical findings, cognitive or developmental disabilities, and the presence of benign tumors ( Table 42-2 ). Primary physicians and pediatric oncologists should be alert to the presence of these manifestations in their patients. If suspicion of a hereditary cancer syndrome is raised, the child should be referred to a geneticist or genetic counselor, as well as appropriate specialist(s) for evaluation, genetic testing, and management.
| Manifestation | Predisposition Syndrome | Gene(s) |
|---|---|---|
| Cutaneous | ||
| Café-au-lait macules | Neurofibromatosis type 1 (NF1) | NF1 |
| Constitutional mismatch repair–deficiency syndrome (CMMR-D) | MLH1, MSH2, MSH6, PMS2 | |
| Axillary or inguinal freckling | NF1 | NF1 |
| Penile freckling | PTEN hamartoma tumor syndrome (PHTS) | PTEN |
| Mucosal pigmentation | Peutz-Jeghers syndrome (PJS) | STK11 |
| Plantar or palmar pits | Nevoid basal cell carcinoma syndrome (NBCCS) | PTCH1 |
| Pilomatrixomas | Familial adenomatous polyposis (FAP) | APC |
| Dental anomalies | ||
| Extra or missing teeth | FAP | APC |
| Jaw osteomas | FAP | APC |
| Jaw keratocysts | NBCCS | PTCH1 |
| Ear anomalies | ||
| Earlobe creases or pits | Beckwith-Wiedemann syndrome (BWS) | 11p15 abnormalities, CDKN1C |
| Eye abnormalities | ||
| Aniridia | Wilms tumor (WT), aniridia, genitourinary malformations, retardation syndrome (WAGR) | WT1 |
| Cataracts | Neurofibromatosis type 2 (NF2) | PTCH1 |
| NBCCS | NF1 | |
| Lisch nodules | NF1 | NF2 |
| Congenital hypertrophy of retinal pigmented epithelium (CHRPE) | FAP | APC |
| Strabismus or leukocoria | Hereditary retinoblastoma (RB) | RB1 |
| Genitourinary anomalies Hypospadias, undescended testes, undermasculinized male external genitals, streak gonads | WT1 -related WT syndromes | WT1 |
| Kidney abnormalities Horseshoe kidney Nephroblastomatosis | WT1 -related WT syndromes | WT1 |
| Overgrowth | ||
| Macrosomia | BWS | 11p15 abnormalities, CDKN1C |
| Simpson Golabi Behmel syndrome | GPC3, CXORF5 | |
| NBCCS | PTCH1 | |
| Macrocephaly | PHTS | PTEN |
| Macroglossia | BWS | 11p15 abnormalities, CDKN1C |
| Hemihypertrophy | BWS | 11p15 abnormalities, CDKN1C |
| Marfanoid habitus | Multiple endocrine neoplasia type 2B | RET |
| Neurologic issues | ||
| Ataxia | Ataxia-telangiectasia | ATM |
| Autism-spectrum disorders | PHTS | PTEN |
| Cognitive delays | WAGR | WT1 |
| NF1 | NF1 | |
| NF2 | NF2 | |
| Seizures | NF1 | NF1 |
| NF2 | NF2 |
Dermatologic Findings
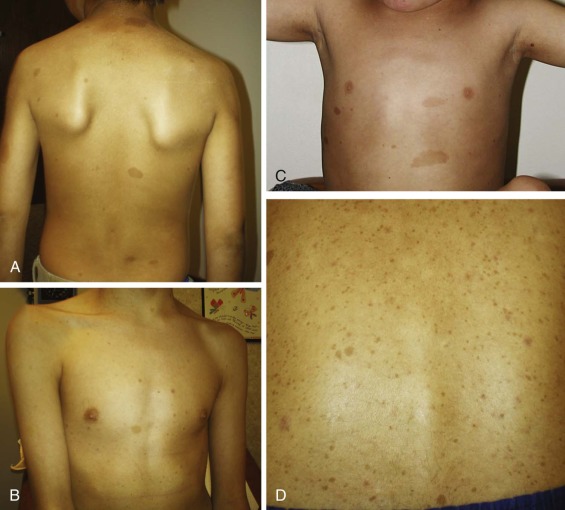
Benign or malignant skin findings are a common feature of several cancer predisposition syndromes, and evaluation by a dermatologist may be helpful in characterizing these findings and facilitating the diagnosis. Café-au-lait macules and axillary and inguinal freckling are a well-known example and are crucial in establishing a clinical diagnosis of neurofibromatosis type 1 (NF1) ( Fig. 42-1 ). Café-au-lait macules are often the earliest clinical manifestation of NF1, as they are present in 99% of affected children by 1 year of age. Café-au-lait macules are also a cardinal feature of CMMR-D. Skin pigmentation differences may also provide a clue to the diagnosis of PTEN hamartoma tumor syndrome (PHTS). For example, boys with PHTS may exhibit pigmented macules of the glans penis, which should raise the suspicion of the diagnosis, especially if features such as macrocephaly, developmental delay, or autism are present. Other skin findings are also found in PHTS, including trichilemmomas and papillomatous papules; however, these may not be diagnosed until later in life. Individuals with Peutz-Jeghers syndrome (PJS) commonly exhibit mucocutaneous pigmentation resembling “freckling” around the mouth, eyes, or nostrils, or on the buccal mucosa or perianal area. Skin cancers in children may also indicate the presence of a syndrome predisposing to cancer. NBCCS predisposes individuals to basal cell carcinomas, which generally appear in the late teens or early adult years, but can occasionally develop in young children. Benign skin findings such as palmar and plantar pits and facial milia may be present in individuals with NBCCS.
Developmental or Cognitive Abnormalities
Developmental or cognitive disabilities, including delays in achieving motor milestones, intellectual disabilities, and autism-spectrum disorders are commonly seen in cancer predisposition syndromes, including PHTS; WT, aniridia, genitourinary malformations, and retardation (WAGR) syndrome; tuberous sclerosis; 13q deletion syndrome (which predisposes to RB); and NF1. Seizures may also be a feature, where they are often seen in NF1 and tuberous sclerosis, and may indicate the presence of an underlying tumor. Any child with cancer who has a history of developmental or intellectual disabilities, autism-spectrum disorders, seizures, and/or other neurologic features should be considered a candidate for referral for a possible cancer genetics evaluation.
Overgrowth
Generalized or focal overgrowth of specific parts of the body can be a feature of a condition predisposing to cancer, such as Beckwith-Wiedemann syndrome (BWS), PHTS, and Simpson Golabi Behmel syndrome. In BWS overgrowth may manifest with generalized macrosomia at birth, and some patients may also exhibit macroglossia and/or asymmetric overgrowth of one side of the body (hemihypertrophy). Hemihypertrophy may involve the extremities or external physical features (face, labia, buttocks), as well as internal organs (where, for example, one kidney may be larger than the other). Macrosomia is also a feature of Simpson Golabi Behmel syndrome and Proteus syndrome (PS), while macrocephaly can be a manifestation of PHTS and NBCCS.
Specific Congenital Anomalies
In some cases, the presence of specific congenital anomalies can serve as an early indicator of a condition predisposing to cancer. For example, an omphalocele or umbilical hernia, especially in a child with macrosomia, macroglossia, ear pits or creases, or neonatal hypoglycemia, can point to the diagnosis of BWS. Another good example is WAGR syndrome, where affected children have aniridia, often in combination with genitourinary abnormalities such as hypospadias or cryptorchidism.
Other Physical Features
Other physical manifestations may also suggest an underlying hereditary cancer syndrome. Examples include individuals with multiple endocrine neoplasia type 2B (MEN2B), who often present with mucosal neuromas of the lips and tongue, ganglioneuromas, and marfanoid habitus, and those with FAP, who may present with dental abnormalities (missing or extra teeth), epidermal cysts or pilomatrixomas, Gardner fibromas (also known as desmoid tumors), osteomas (usually involving the jaws), and congenital hypertrophy of the retinal pigmented epithelium. Generally these manifestations present prior to the development of colon polyps.
Incidental Identification of a Child with a Condition Predisposing to Cancer
Until recently, genetic testing for hereditary cancer syndromes was only offered when an individual presented with a specific cancer type, physical or developmental features consistent with a known syndrome predisposing to cancer, or a suggestive family history. Due to the increasing use of genome-wide approaches to establish a genetic diagnosis, including single-nucleotide polymorphism arrays, multigene panels, and whole-exome sequencing, the “incidental” discovery of a gene mutation that predisposes to cancer is becoming more and more common. This information can lead to the unintended diagnosis of a predisposing syndrome prior to its clinical manifestation in the child and/or his or her family. Toward this end, several recent studies of genome-wide microarrays have reported the identification of copy number variations involving genes predisposing to cancer in individuals presenting with a noncancer phenotype. In these studies, the frequency of incidental findings involving genes predisposing to cancer was estimated to be between 0.18% and 0.60%.
The testing of tumor samples for somatic genetic mutations is also being pursued with the intent of facilitating “personalized” cancer care by identifying and therapeutically targeting mutated genetic pathways in tumors. When evaluating tumors, it is often necessary to compare tumor-derived sequences to those obtained using normal tissue from the same individual. As a result, it is possible to identify incidental constitutional mutations that are indicative of an underlying cancer predisposition or other nononcologic condition. In one recent study, most patients undergoing tumor genetic testing expressed interest in receiving incidental germline genetic test results, as most thought this information would be valuable to family members. However, some perceived it to be an additional burden on top of their cancer diagnosis. Many laboratories have specific policies regarding the sequencing of normal tissue and return of incidental results. Therefore, prior to ordering tumor testing, pediatric oncologists should be familiar with the performing laboratory’s policy, discuss the possibility of obtaining incidental genetic findings with the patient and his or her parents, and determine whether or not they would like to receive these results as part of the informed-consent process.
Genetic Testing for a Heritable Predisposition to Cancer
Complexities of Childhood Cancer Genetic Testing
Genetic testing for cancer predisposition is a complex process that should be explored with the patient and family, preferably by a genetic counselor or trained genetics professional, prior to performing the testing. Counseling should include information about the natural history of the condition under consideration, methods of testing and its possible outcomes, as well as the potential risks, benefits, and limitations of testing. Patients may have concerns about privacy, confidentiality, insurance coverage, and genetic discrimination, all of which must be addressed. For the oncologist, it is important to recognize that genetic test results have implications not only for the individual being tested but also for his or her family members. This factor necessitates counseling of the whole family and discussion of how the results will be communicated between various members. When the individual under consideration is a minor (defined here as a person <18 years), the importance of these issues is further magnified by the fact that children generally do not have the capacity to fully understand the risks, benefits, and limitations of genetic testing. Therefore, they are not able to make their own informed decisions about whether or not to be tested. Instead, this decision falls to the child’s parent(s) or guardian(s), and strips the child of the opportunity to make an autonomous decision about genetic testing later in life. Testing in childhood and disclosure of results to parents, guardians, and/or other family members may violate the child’s privacy, confidentiality, and right not to know. Many also fear that performing genetic testing on a child may have adverse psychological consequences, such as depression, anxiety, fear of the future, altered self-image, limited horizons (i.e., impact on thoughts about future education and career choices), and altered family relationships. However, others have argued for the possible benefits of genetic testing in childhood, such as relief if test results are negative, providing certainty about genetic risk, and allowing the child to integrate genetic information into the self-concept at a younger age, when it is less disruptive to the sense of self. Until recently, the concerns about testing children have more or less superseded the possible benefits of such testing.
Despite much debate, available evidence suggests that the negative consequences of cancer genetic testing may be fewer and less severe than anticipated. Studies of the psychosocial consequences of genetic testing in adults have generally shown minimal or no negative consequences and suggested that emotional outcomes may be more dependent on factors such as pretest emotional state and social support than on the actual genetic test result itself. Several studies report that adults undergoing genetic testing for LFS consider obtaining certainty about cancer risk a psychological benefit of testing, even if adults are found to be affected. Data on the outcomes of presymptomatic cancer genetic testing of children are more limited. Much of the data comes from testing for FAP, where affected individuals are prone to develop multiple gastrointestinal polyps, which if left untreated invariably lead to early-onset colorectal cancer. In FAP, polyps generally begin to form during adolescence. Consequently, it is recommended that children begin surveillance with colonoscopy around the age of 10 to 12 years. Based on these factors, it is deemed appropriate to offer FAP genetic testing to minors. Published reports describing the psychosocial consequences of pediatric FAP genetic testing reveal that children by and large do not experience significant distress related to this testing. One report describing the psychological outcomes of genetic testing of minors for LFS did not note any significant negative outcomes after up to 12 years of follow-up. However, in this study, the participation rate in testing for minors was low, with only 4 of 26 parents who were offered testing for their children actually following through with the test. Therefore, it remains to be determined whether and how genetic testing for these or other conditions predisposing to cancer impacts the well-being of children and their families.
Guidelines for Testing Children for a Heritable Predisposition to Cancer
The controversies surrounding genetic testing of children have prompted several groups to issue guidelines and recommendations. Most recommendations indicate that the primary justification for presymptomatic (i.e., predictive) genetic testing is medical benefit for the child. Therefore, testing should be offered to minors only if the condition is known to manifest in childhood and there are effective preventive or therapeutic interventions available. Conversely, if cancer risk is not increased in childhood or if there are not effective surveillance or intervention measures, it is recommended that testing be delayed until adulthood, when a child can then independently decide whether or not to pursue the testing. Families must be counseled about the benefits, risks, and limitations of genetic testing in order to give informed consent for genetic testing on behalf of their child. As with other medical interventions, it is the role of the health care provider to advocate for the best interests of the child when discussing the option of genetic testing. If possible, minors should be encouraged to participate in decision making about genetic testing; however, the capacity of children to understand genetic information is variable and depends upon their age, maturity, education, and cognitive abilities. Disclosure of results to tested minors is also encouraged once the minor reaches the legal age of majority, or possibly sooner if it is felt that the minor is capable of understanding and coping with the results.
Management of Children with a Heritable Predisposition to Cancer
The identification of a presymptomatic child with a heritable predisposition to cancer has the potential to improve survival and minimize morbidity by allowing prospective monitoring for tumor formation and often life-saving prophylactic surgeries or other preventive measures. This information can also benefit children who already have cancer, as it can influence choice of the most appropriate or least toxic cancer-directed therapy. For example, one should avoid the use of radiation therapy in children with hereditary RB or LFS, as these children are at increased risk of developing secondary irradiation-induced malignant disease due to their constitutional genetic mutations. Similarly, in a child with a WT predisposition syndrome, it is recommended that a renal tumor be removed using nephron-sparing surgery and not total nephrectomy. Because the remaining kidney tissue remains at risk for tumor formation and could thus become the target of future surgical procedures, it is deemed advisable to leave as much healthy kidney in place as possible. Below, we review some of the salient principles of tumor surveillance and cancer risk reduction for children with heritable predisposition to cancer.
Principles of Tumor Surveillance
Cancer surveillance guidelines exist or are being developed for several conditions predisposing to childhood cancer. The primary goal of surveillance is to detect cancers at the earliest and most curable stage. For this reason, cancer surveillance protocols are best suited for solid tumors, where survival is often linked to the size and extent of spread of the tumor at diagnosis. Tumors identified in individuals undergoing regular monitoring may be smaller. As a result, surveillance allows for less invasive surgical procedures and can also reduce or eliminate the need for additional chemotherapy and/or irradiation. However, surveillance can also be complicated by the possibility of false-positive results, which often lead to worry about cancer, excess rates of follow-up imaging and/or biopsy, and possibly increased costs of care.
Several factors must be taken into account in the development and implementation of a cancer surveillance protocol. First, there must be a benefit for early cancer detection in an individual undergoing monitoring (i.e., there should be an effective treatment available and preferably also demonstration that early detection improves outcome). Second, the age-specific tumor risks for syndromes predisposing to cancer must be known and deemed high enough to warrant surveillance (generally, a tumor incidence of ≥5% is considered sufficiently high to initiate monitoring). This information helps to identify candidates for surveillance, when it should be initiated, and how long it should be continued. The surveillance method(s) and interval between specific tests must also be carefully considered in light of the specific tumor risks and growth rate associated with a given syndrome. Ideally, surveillance methods should be readily available, safe, and have high sensitivity and specificity. Whenever possible, screening tools that involve no or minimal irradiation should be used, as patients with hereditary cancer syndromes may be at increased risk to develop irradiation-induced cancers (as is the case for hereditary RB, LFS, ataxia telangiectasia, and other genetic syndromes associated with defects in DNA repair).
Cancer Prevention Strategies
For some syndromes, early identification of at-risk children allows for the elimination or dramatic reduction of cancer risk through prophylactic surgery. MEN2 and FAP are excellent examples, where removal of the at-risk organ may be considered early in a patient’s lifetime to prevent the development of thyroid or colorectal cancer, respectively. In MEN2, there is a nearly 100% lifetime risk for MTC, an aggressive type of thyroid cancer with a high metastatic potential. As a result, it is recommended that individuals with MEN2 undergo prophylactic thyroidectomy, a procedure that has greatly reduced the likelihood of developing MTC. It should be noted that while all individuals with MEN2 are at increased risk for MTC, there are significant genotype-phenotype correlations that predict the age of thyroid cancer onset, and this information can dictate the appropriate timing for prophylactic removal of the thyroid. In FAP, individuals have a similarly high risk of early-onset colon cancer that nears 100% if colonic polyps are not removed. To prevent colon cancer, affected individuals should undergo colectomy when polyps become too numerous to clinically follow or remove, generally in the late teens to early 20s. Studies of the efficacy of colectomy reveal a significant reduction in colon cancer risk. Due to the morbidities associated with colectomy, alternatives to surgery are being sought. Nonsteroidal antiinflammatory medications such as sulindac and Cox-2 inhibitors reduce polyp formation in adults with FAP ; however, these medications are currently under examination for use in children with the condition.
Specific Syndromes Predisposing to Cancer
There currently exist more than 30 genetic conditions that predispose to cancer in which children are at risk for tumor formation (several examples are summarized in Table 42-3 ). Each of these conditions is typified by distinct clinical and genetic manifestations. For some of these conditions, recommendations regarding genetic testing, surveillance, and management are well established. However, for others these issues are only recently emerging. Here we focus on some of these conditions, including the clinical and molecular features and current recommendations regarding management.
| Syndrome | Gene(s) | Inheritance | Cancers or Tumors | Other Features |
|---|---|---|---|---|
| Beckwith-Wiedemann syndrome | Chromosome 11p15, CDKN1C | Imprinting defects Autosomal dominant (AD) | Hepatoblastoma (HB) Wilms tumor (WT) Others (rare): Adrenocortical cancer Neuroblastoma Pheochromocytoma (PCC) Rhabdomyosarcoma Rhabdoid tumor Gonadoblastoma | Macrosomia Macroglossia Omphalocele Umbilical hernia Hypoglycemia Hemihypertrophy Ear pits or creases |
| Constitutional mismatch repair–deficiency syndrome | MLH1 MSH2 MSH6 PMS2 | Autosomal recessive (AR) | Gastrointestinal polyps (adenomas) Colon cancer Brain cancer Leukemia or lymphoma Endometrial cancer | Café-au-lait spots Axillary or inguinal freckling Lisch nodules |
| DICER1 syndrome | DICER1 | AD | Pleuropulmonary blastoma Cystic nephroma Sertoli–Leydig cell tumor Thyroid goiter WT Rhabdomyosarcoma Colon polyps (hamartomas) | Pulmonary cysts |
| Familial adenomatous polyposis | APC | AD | Gastrointestinal polyps Colorectal cancer Small bowel cancer Papillary thyroid cancer HB Medulloblastoma Soft tissue tumors Gardner fibroma Desmoid tumor Osteomas | Epidermoid cysts Pilomatrixomas Supernumerary or missing teeth Congenital hypertrophy of retinal pigmented epithelium (CHRPE) |
| Hereditary paraganglioma (PGL)-PCC syndrome | SDHA/B/C/D/AF2 MAX TMEM127 | AD | PCC PGL Papillary thyroid cancer Renal cancer Gastrointestinal stromal tumor (GIST) | |
| Hereditary neuroblastoma | ALK PHOX2B | AD | Neuroblastoma | Hirschsprung disease ( PHOX2B ) Congenital central hypoventilation ( PHOX2B ) |
| Hereditary retinoblastoma | RB1 | AD | Retinoblastoma Pineoblastoma Soft tissue or bone sarcoma Melanoma | Multiple congenital anomalies (13q syndrome) |
| Juvenile polyposis syndrome | BMPR1A SMAD4 | AD | Colon cancer Gastric cancer Pancreatic cancer Gastrointestinal polyps Juvenile type | Arteriovenous malformations ( SMAD4 ) |
| Li-Fraumeni syndrome | TP53 | AD | Adrenocortical carcinoma Choroid plexus carcinoma Bone or soft tissue sarcoma Breast cancer Brain cancer Leukemia Colon cancer Many other cancers | None |
| Multiple endocrine neoplasia type 1 | MEN1 | AD | Parathyroid adenoma Gastrinoma-insulinoma Anterior pituitary adenoma Carcinoid tumor Angiofibroma Collagenoma Lipoma | None |
| Multiple endocrine neoplasia type 2 | RET | AD | Medullary thyroid cancer PCC Parathyroid adenoma (MEN2A) Mucosal neuroma (MEN2B) Ganglioneuroma (MEN2B) | Hyperparathyroid (due to parathyroid adenomas) (MEN2A) Marfanoid habitus (MEN2B) Mucosal neuromas (MEN2B) |
| Neurofibromatosis type 1 | NF1 | AD | Neurofibromas Optic pathway gliomas Astrocytomas Schwannomas Juvenile myelomonocytic leukemia Acute myeloid leukemia PCCs | Café-au-lait macules Axillary or inguinal freckling Lisch nodules Tibial bowing Macrocephaly Developmental delay, intellectual disability, or autism |
| Neurofibromatosis type 2 | NF2 | AD | Vestibular schwannomas Meningiomas Astrocytomas Schwannomas Gliomas Neurofibromas | Posterior subcapsular lenticular opacities Hearing loss Peripheral neuropathy Seizures |
| Nevoid basal cell carcinoma syndrome (Gorlin syndrome) | PTCH1 | AD | Basal cell carcinoma Medulloblastoma Cardiac and ovarian fibromas | Macrocephaly Palmar or plantar pits Jaw keratocysts Bifid ribs Calcificiation of falx Developmental delay, intellectual disability, autism |
| Peutz-Jeghers syndrome | STK11/LKB1 | AD | Gastrointestinal polyps (Peutz-Jeghers type) Colon cancer Gastric cancer Breast cancer Lung cancer Pancreatic cancer Ovarian cancer Cervical cancer | Mucocutaneous hyperpigmentation |
| PTEN hamartoma tumor syndrome | PTEN | AD | Breast cancer Thyroid cancer (nonmedullary) Endometrial cancer Renal cancer Lipomas Thyroid nodules or goiter Gastrointestinal polyps (hamartomas) | Macrocephaly Arteriovascular malformations Trichilemmomas Papillomatous papules Developmental delay, intellectual disability, or autism |
| Rhabdoid tumor syndrome | SMARCB1/INI1 | AD | Atypical teratoid/rhabdoid tumors (CNS) Malignant rhabdoid tumors (renal or extrarenal) Schwannomatosis | None |
| Tuberous sclerosis | TSC1 TSC2 | AD | Astrocytoma Cortical tuber Subependymal nodules Cardiac rhabdomyoma Angiomyolipomas Renal cell carcinoma (RCC) Colon polyps (hamartomas) | Facial angiofibroma Hypopigmented macules (ash leaf macules) Ungual fibromas Shagreen patch Confetti skin lesions Dental pits Gingival fibromas Developmental delay, intellectual disability, or autism Seizures or infantile spasms |
| Von–Hippel Lindau syndrome | VHL | AD | Hemangioblastoma (retinal or cerebellar) RCC Pancreatic neuroendocrine tumors PCCs Endolymphatic sac tumors Papillary cystadenomas Epididymis (males) Broad ligament (females) | Renal cysts Pancreatic cysts |
| WT1 -related syndromes | WT1 | AD | WT Gonadoblastoma | Aniridia (WAGR syndrome) Genitourinary abnormalities Developmental delay, intellectual disability, or autism |
Hereditary Retinoblastoma
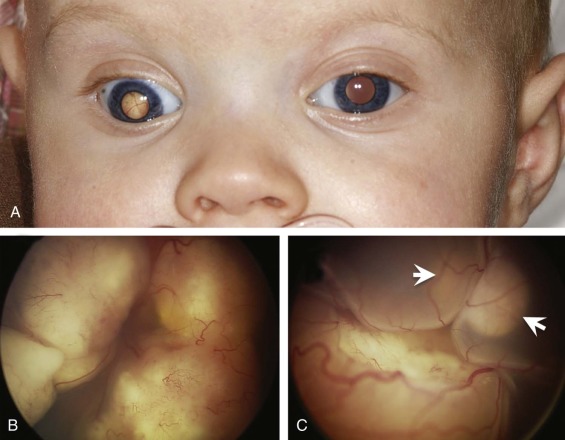
Incidence
Hereditary RB is an example of a classic syndrome predisposing to cancer and the first for which the underlying genetic mechanism was recognized. RB is a malignant tumor of the embryonic neural retina that has an incidence of two to five cases per million children per year and accounts for 3% to 4% of all pediatric cancers. Nonhereditary RB accounts for 60% of cases and is associated with development of unilateral eye tumors at a median age of 22 months. The remaining 40% of patients have hereditary RB, which involves both eyes in more than 80% of cases and presents at a median age of 11 months. Heritable RB is highly penetrant, with more than 90% of patients developing RB and 0.5% to 15% also developing intracranial tumors of the pineal gland. Over the course of their lives, survivors of hereditary RB remain at increased risk to develop second malignant neoplasms, including bone and soft tissue sarcomas, which commonly occur in previously irradiated sites, as well as melanomas and cancer of the lungs, gastrointestinal tract, and bladder.
Genetics
RB is caused by mutations in RB1 , which resides at chromosomal locus 13q14. RB1 encodes the RB protein (pRB), a potent tumor suppressor that regulates cell growth by inhibiting the function of the E2F family of transcription factors and preventing inappropriate S-phase entry. pRB also regulates cellular glucose tolerance, mitogenesis, glutathione synthesis, and the expression of genes involved in carbon metabolism. When both RB1 alleles are mutated within a developing retinal cell, pRB expression and/or function are diminished, cell division proceeds in a dysregulated manner, and, ultimately, RB tumors form.
In patients with nonhereditary RB, both RB1 gene copies are mutated within a single postzygotic retinal cell. In contrast, patients with hereditary RB harbor one mutated RB1 allele within the germline. This mutation is inherited from a similarly affected parent in 10% to 20% of cases. In the remaining cases, the RB1 mutation arises de novo within one of the parent’s gametes or a cell of the developing embryo. Mutation of the remaining RB1 allele occurs somatically. Genetic testing for germline RB1 gene mutations can identify mutations in more than 90% of individuals with hereditary disease. While most patients can be identified clinically due to the presence of multifocal or bilateral eye tumors and/or a positive family history, 10% to 15% of patients have single eye tumors and no family history of RB. These latter factors make them indistinguishable from those with the sporadic form of disease. The identification of RB1 mutations in unilateral patients enables appropriate primary eye tumor treatment as well as initiation of surveillance for involvement of the pineal gland and later second cancers. Identification of a germline RB1 mutation in any child with RB also allows for presymptomatic testing of other family members, including siblings who may not yet have developed RB but carry a mutated RB1 gene copy, as well as children of RB survivors.
Tumor Risks and Surveillance
Individuals harboring germline RB1 mutations are at greatly increased risk to develop bilateral RB tumors, generally during the first 5 years of life ( Fig. 42-2 ). During this time period patients are also at increased risk to develop tumors involving the pineal gland (so-called trilateral RB). Later in life, individuals with hereditary RB can develop second malignant neoplasms. While the risk for these cancers is greatest in those treated with external-beam radiation therapy, nonirradiated long-term survivors of hereditary RB have an approximately 10% to 15% risk of developing one or more second malignant neoplasms