Genetic Defects Causing Hypothyroidism
Thomas Vulsma
Jan J.M. De vijlder
Congenital hypothyroidism (CH), a lack of thyroid hormone (TH) already present at birth, is a relatively common medical condition with large variations in severity, duration, etiology, clinical expression, and outcome. TH is needed for almost all physiologic functions in the human body, and above all of crucial importance for brain development. The risk of irreversible brain damage mainly depends on severity and duration of the TH deficiency. Since maternal-fetal transfer of thyroxine (T4) partly compensates for insufficient fetal T4 production (1,2), most clinical manifestations of CH are not yet observed at birth (see Chapter 54B).
CH can be caused by endogenous, genetic defects, or by exogenous disruptors, originating from the maternal environment. In general, genetic defects cause permanent
hypothyroidism, and exogenous influences provoke transient hypothyroidism, but there are important exceptions. Exogenous disruptions of the fetus’ or neonate’s thyroid function are, at least in principle, predictable and often preventable events, whereas genetic defects causing hypothyroidism can neither be prevented, nor cured. Fortunately, the severe adverse effects of lack of T4 on brain development can be largely prevented by early postnatal treatment with T4 (3). Consequently, neonatal mass screening programs for CH are indispensable (see Chapter 54A).
hypothyroidism, and exogenous influences provoke transient hypothyroidism, but there are important exceptions. Exogenous disruptions of the fetus’ or neonate’s thyroid function are, at least in principle, predictable and often preventable events, whereas genetic defects causing hypothyroidism can neither be prevented, nor cured. Fortunately, the severe adverse effects of lack of T4 on brain development can be largely prevented by early postnatal treatment with T4 (3). Consequently, neonatal mass screening programs for CH are indispensable (see Chapter 54A).
The spectrum of genetic defects causing hypothyroidism in humans comprises defects in the development of thyroid, pituitary, or hypothalamus, in the synthesis of T4, or TSH, in the TH cell membrane transporters, or TH nuclear receptors in the peripheral (target) tissues, and in the recycling of iodine (dehalogenation of iodotyrosines) in both the thyroid and peripheral organs. Finally, CH can be part of a number of congenital syndromes.
Neonatal screening has increased the need for etiologic classification to verify the diagnosis, to optimize the treatment, and to enable genetic counseling. To obtain an accurate clinico-pathologic diagnosis, sensitive immunoassays for the various diagnostic determinants of the thyroid function, and thyroid ultrasound imaging are required. These are (when indicated) combined with in vivo radioiodine (123I) studies, analysis of the iodinated metabolites excreted in urine, brain magnetic resonance imaging (MRI), thyrotropin-releasing hormone (TRH) stimulation testing, or stimulation tests for the other pituitary functions. With these tools, information is obtained about the gland(s) involved, gland morphogenesis, the risk of goitrogenesis and nodular degeneration, the likelihood of transient hypothyroidism, the risk of involvement of other hormone deficiencies, or defects in other organ systems (Table 36.1). Finally, the putative etiology may point to the gene, or a selection of candidate genes that may harbor the causal mutation(s).
In practice, the contributions of molecular biology to the etiologic classification of CH are progressively replacing in vitro studies of tissues (4,5,6). However, it should be kept in mind that it is not always possible to establish whether a gene mutation is causally related to CH. People may have mutations that theoretically influence thyroid function, whereas they turn out to be and remain euthyroid (7). Finally, most cases of well-established permanent CH still lack a molecular classification.
It is not possible to establish accurate cut-off levels for blood or serum (free) T4 or TSH that discriminate between normal and CH, and neither there exist cut-off levels for the risk of brain damage. Therefore, worldwide, screening cut-off levels have been chosen empirically, aiming to minimize the risk of missing (even mild) cases of CH, and to keep unnecessary referrals as low as possible. As a result, the introduction of neonatal CH screening programs has given rise to considerably increased (apparent) incidence figures, mainly because of the referral of cases with mildly elevated TSH concentrations. Nevertheless, further lowering the cut-off level of TSH up to the upper limit of the reference range has shown to increase the apparent CH incidence figures considerably (8). However, the clinical relevance of the early detection of (very) mild cases of CH has yet to be established. Indeed, mild CH was virtually unknown in the prescreening era, and there is a lack of placebo controlled studies to the outcome of early started T4 supplementation (9). One of the main reasons for etiologic classification in milder cases of CH is to warrant that at least the diagnosis is beyond doubt.
Until now, almost all screening programs worldwide only detect CH of thyroidal origin (CH-T). Indeed, approximately 90% of the newborn infants with CH, detected by the Dutch screening program (based on measurement of T4 with additional measurements of TSH and thyroxine-binding globulin) have CH-T, either due to a developmental defect (thyroid dysgenesis, 80%), or a defect in the biosynthesis of T4 (thyroid dyshormonogenesis, 10%). The remaining 10% of the patients diagnosed by the Dutch approach have CH of central origin (CH-C) (10,11). A delayed diagnosis in these cases may have major implications for morbidity and mortality, especially if there is also concomitant cortisol deficiency caused by the hypothalamic or pituitary defect.
Central Congenital Hypothyroidism
The apparent incidence of CH-C in The Netherlands, at least 1:20,000 newborn infants, as obtained from Dutch screening program, is substantially higher than usually assumed (10,12,13). Recent data from a region in Japan confirm this observation (14).
Like CH-T, also CH-C includes a heterogeneous group of disorders. Commonly, the TSH deficiency occurs with other multiple pituitary hormone deficiencies (MPHD). Although most cases are sporadic, they presumably originate from genetic defects. For unknown reasons, CH-C is more common in males than females (13), although a small percentage of cases with MPHD or isolated CH-C can be ascribed to mutated genes located on the X chromosome (15) (unpublished observations). Many infants with CH-C, especially those with MPHD, have a developmental abnormality of the diencephalic infundibulum, with the posterior lobe of the pituitary localized at the median eminence, just below the floor of the third ventricle. Such an ectopic posterior pituitary (EPP) is usually visible as a characteristic “bright spot” on T1-weighted MRI.
Defects in Ontogeny of Hypothalamus and Pituitary Gland
The hypothalamus develops from the infundibulum, a region of the ventral diencephalon (see Chapter 3). It contains several nuclei that produce releasing and inhibiting hormones that regulate synthesis and secretion of the various pituitary hormones. Defective neural migration in the developing infundibulum, resulting in EPP, is often accompanied by other malformations in the brain (e.g., periventricular heterotopia) (13,16) or elsewhere, and the functional problem may vary from isolated growth hormone (GH) deficiency to panhypopituitarism. In a few patients, mutations in the transcription factor genes HESX1 and OTX2 have been associated with this phenotype (17).
The pituitary gland develops from two distinct structures, both of ectodermal origin (see Chapter 2). The posterior lobe develops from the infundibulum, and the intermediate and anterior lobes derive from Rathke’s pouch. Mutations in a series of homeobox genes, including HESX1, LHX3, LHX4, PHF6, SOX2, SOX3, OTX2, PITX1, PITX2, PROP1, and
POU1F1 may result in various types of congenital hypopituitarism (18). The genetic defects responsible for the various pituitary anomalies can be classified roughly into three categories: (I) mutations in genes involved in early embryonic development (often structures adjacent or more distant to the pituitary may be affected too), (II) mutations in genes involved in initial stages of pituitary cell differentiation (mostly resulting in MPHD), (III) mutations in genes involved in production and secretion of the pituitary’s stimulating hormones. In this section only the genetic defects that carry the risk of CH-C will be considered.
POU1F1 may result in various types of congenital hypopituitarism (18). The genetic defects responsible for the various pituitary anomalies can be classified roughly into three categories: (I) mutations in genes involved in early embryonic development (often structures adjacent or more distant to the pituitary may be affected too), (II) mutations in genes involved in initial stages of pituitary cell differentiation (mostly resulting in MPHD), (III) mutations in genes involved in production and secretion of the pituitary’s stimulating hormones. In this section only the genetic defects that carry the risk of CH-C will be considered.
Table 36.1 Etiologic Classification of Genetic Defects Causing Hypothyroidism | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Mutations in Hesx1 in mice cause defects comparable with septo-optic dysplasia in humans (19). This entity is characterized by optic nerve hypoplasia, various forebrain defects (often including corpus callosum and septum pellucidum defects), and MPHD (often including TSH deficiency). Inheritance can be dominant or recessive (17,20,21). Eight homozygous mutations in LHX3 have been described in 13 patients from 8 unrelated consanguineous families. Most patients had severe deficiencies of all anterior pituitary hormones, except adrenocorticotropic hormone (ACTH), and had a rigid cervical spine that limited head rotation (presumably related to the role of LHX3 in motor neuron development) (22). Recently a patient with septo-optic dysplasia and homozygous mutations in LHX3 has been described who also had ACTH deficiency (23).
In a family with short stature, pituitary and cerebellar defects, poorly developed sella turcica, and MPHD (including TSH deficiency) a mutation was found in the LHX4 gene that abolished normal splicing and delivered either a protein with deletion of four amino acids within the homeobox domain, or a truncated protein. The aberrant LHX4 gene products were unable to bind to a putative binding site within the POU1F1 promoter due to decreased DNA binding (24). The inheritance was autosomal dominant.
The transcription factor SOX2 is able to bind to sequences within the promoter of HESX1 and LHX3, which suggests that it might be a regulator of pituitary development as well. Mutations in this gene are associated with variable hypopituitarism and midline defects (17).
The Börjeson–Forssman–Lehmann syndrome is a rare X-linked condition, characterized by early onset of MPHD (most affected males are short and have hypogonadism), optic nerve hypoplasia, mild-to-moderate intellectual handicap, epilepsy, and gradual development of coarse facial features. Heterozygous females are variably affected (25,26). This syndrome has been ascribed to mutations in PHF6 (27,28). The clinical features suggest that it plays an important role in midline neurodevelopment, including the hypothalamus and the pituitary gland. There is a case report of a boy with a Börjeson-Forssman-Lehmann-like syndrome who had a duplication in chromosome Xq26.3-q28 (29).
A number of pedigrees with X-linked-recessive hypopituitarism and mild-to-moderate mental retardation caused by chromosomal duplications of Xq26.1-q27.3 (28,29), encompassing SOX3 have been described (30). The phenotypes of affected males involving duplications within this region are variable. They were all GH deficient and showed hypoplasia of the anterior pituitary, infundibular hypoplasia and EPP. Moreover abnormalities of the corpus callosum were observed (31), and deficiencies of ACTH, TSH, or panhypopituitarism have been described (32). Polyalanine tracks have been identified within SOX3, and expansion of this polyalanine track with 11 alanine residues (33) resulted in GH deficiency and mental retardation. In three siblings of a consanguineous pedigree, an expansion of 7 alanine residues gave profound and complete panhypopituitarism, in association with anterior pituitary hypoplasia and EPP (30). A deletion of the same repeat by 9 alanine residues was found in two brothers with mental retardation. However, since the same deletion was found in the clinically unaffected grandfather, it is uncertain whether this deletion is causal (33).
A patient with bilateral anophthalmia and panhypopituitarism, associated with a small anterior pituitary lobe, EPP, absent infundibulum and Chiari malformation, had a heterozygous de novo 2-bp insertion in exon 3 of the OTX2 gene (34). This insertion created a protein lacking a C-terminal transactivation domain, by which it failed to activate the promoters of HESX1 and POU1F1. A frameshift mutation in the OTX2 gene was detected in a Japanese woman with short stature, bilateral anophthalmia and a normal anterior pituitary lobe by MRI. The truncated protein was associated with reduced transactivation at the HESX1 and POU1F1 promoters (35). A heterozygous point mutation in the C-terminal transactivation domain of the OTX2 gene (Asn233Ser) in two unrelated children with MPHD gave rise to reduced transcriptional activation properties, and acted as well as a dominant-negative inhibitor of the HESX1 promoter (36).
PROP1 mutations are the most common genetic cause of MPHD, accounting for approximately 50% of familial cases (17). The disorder is recessively inherited; families with homozygosity or compound heterozygosity for inactivating point mutations and deletions in the PROP1 gene have been described (37). The mutation is often a 2-bp deletion in an AG-rich region in exon 2, resulting in a premature stop codon; another mutation (149delGA) resulting in the same termination has been described. To date 26 distinctive mutations have been identified in more than 180 patients. The hormonal deficiencies in patients with inactivating mutations in PROP1 emerge in a typical order. Neonatal features of hypothyroidism, such as prolonged jaundice have been described only once, and growth appears to be normal during infancy (38,39). GH deficiency becomes evident before TSH deficiency in 80% of patients, and gonadotropin deficiency develops later, although most patients fail to enter puberty spontaneously. ACTH deficiency may not be manifest until several decades after birth. The degree of prolactin deficiency is variable. Although all untreated patients end up extremely short and sexually immature, their intelligence is normal, suggesting that they had been euthyroid during the first few years after birth.
Pituitary morphology in patients with PROP1 deficiency varies from hypoplastic to hyperplastic, without involvement of the surrounding tissues. The distinct MRI characteristics of pituitary enlargement may originate from the intermediate lobe (40).
Developmental defects such as solitary maxillary central incisor, nasal pyriform aperture stenosis, and variants of holoprosencephaly can be clues to the presence of deficiencies of one or more pituitary hormones (41).
The transcription factor POU1F1 acts as one of the main stimulators of TSH, GH, and prolactin synthesis. Heterozygous, compound heterozygous, and homozygous POU1F1
deletions, and missense and nonsense mutations have been reported (42); the phenotype varies even between siblings with the same mutation (42,43,44,45,46). A heterozygous Arg271Try mutation exerts a dominant-negative effect (47). Hypothyroidism may be present at birth and has been detected in the Dutch CH screening program.
deletions, and missense and nonsense mutations have been reported (42); the phenotype varies even between siblings with the same mutation (42,43,44,45,46). A heterozygous Arg271Try mutation exerts a dominant-negative effect (47). Hypothyroidism may be present at birth and has been detected in the Dutch CH screening program.
Holoprosencephaly, the most common forebrain defect in humans, is the result of incomplete midline cleavage of the prosencephalon. A number of genes (e.g., SHH, ZIC2, SIX3, GLI2) are involved, but there is often little correlation between phenotype and genotype. Presumably, congenital hypopituitarism is not uncommon in the more severe types of holoprosencephaly, although pituitary function has rarely been described in detail. Posterior pituitary function seems more often affected than anterior pituitary function; CH-C has been reported to occur in 11% (48). A solitary median maxillary central incisor has been reported in holoprosencephaly and in association with a number of other congenital defects, including hypopituitarism. This dental abnormality is associated with mutations in SHH and SIX3 (49).
Defects in Synthesis of Thyrotropin
TRH, the tripeptide pyroglutamyl-histidyl-prolineamide, is the major hypothalamic mediator of the synthesis and secretion of TSH. It is present throughout the brain, but is found in highest concentrations in the paraventricular nuclei and median eminence of the hypothalamus (see Chapter 10C). Until now, no patient with a mutation in the gene for the precursor of TRH (preproTRH) or the gene for the enzymes that cleave prepro-TRH to its component TRH molecules has been described.
Although the TRH receptor on the cell membrane of the TSH producing cells in the pituitary is essential for maintaining a normal thyroid function, some production and secretion of TSH remains, even in patients with biallelic mutations that completely inactivate the TRH receptor (50). Patients with this type of CH-C have moderate or mild hypothyroidism; the absence of any response on secretion of TSH (and prolactin) after administration of exogenous TRH confirms complete resistance to TRH.
TSH stimulates growth and function of the thyroid via interaction with its specific plasma membrane receptor (TSHR). Genetic disorders of TSH synthesis are rare. They include patients whose TSH has decreased bioactivity, resulting in mild hypothyroidism and high serum concentrations of immunoreactive TSH as well as patients with totally inactive TSH, thyroid gland hypoplasia, and severe CH (51). In the latter condition, basal serum TSH concentrations are extremely low, not responding on administration of exogenous TRH, while basal and stimulated prolactin levels are normal.
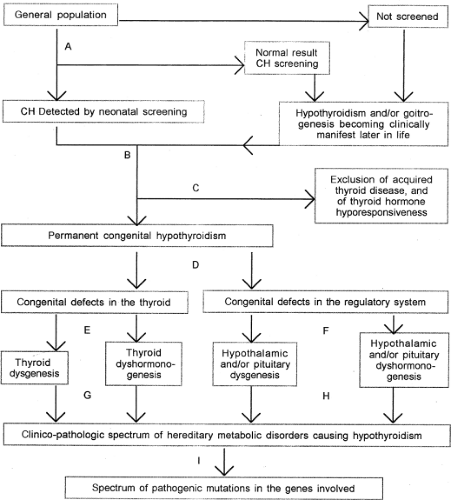 Figure 36.1 Flow chart for etiologic classification of disorders causing congenital hypothyroidism (CH). A: Neonatal screening by measurements of thyroxine (T4) and thyrotropin (TSH) in dried blood samples on filter paper. B: Confirmation by measurement of serum TSH and serum free T4. C: If the serum free T4 concentration is inappropriately high in relation to the serum TSH concentration, clinical evaluation of the infant and measurement of maternal TSH receptor-stimulating antibodies and urinary iodine excretion are indicated. D: Decide whether the serum free T4 and TSH values indicate the presence of thyroidal or central CH. E: Perform thyroid radionuclide imaging or ultrasonography to distinguish between thyroid dysgenesis or thyroid dyshormonogenesis. F: Perform MRI of the hypothalamus and pituitary. G: Measure serum thyroglobulin and thyroid radioiodide uptake (including discharge after perchlorate), and test urine for iodopeptides or iodotyrosines. H: Evaluate other components of hypothalamic–pituitary function. I: Perform mutation analysis to identify the specific defect. |
TSH is a heterodimer composed of noncovalently linked α- and β-subunits. It shares the α-subunit with follicle-stimulating hormone, luteinizing hormone, and chorionic gonadotropin; each has a different β-subunit. A variety of biallelic inactivating mutations has been described in the gene for the β-subunit of TSH, e.g., mutations that hinder dimerization with the α-subunit (52), mutations that resulted in a truncated inactive TSH (53,54,55,56) and frame shifts (57,58).
Thyroidal Congenital Hypothyroidism
On average, the overall incidence of CH-T is 1:4,000, but it varies considerably in different countries and among different racial and ethnic groups. The majority of patients with CH-T have abnormal thyroid development (thyroid dysgenesis). Therefore, the diagnostic evaluation should include an imaging procedure. If the infant has a normally shaped and located thyroid gland, irrespective of its size, and a high serum TSH concentration, further studies with radioiodide will provide information about thyroidal iodide metabolism. Measurements of serum thyroglobulin and low-molecular-weight iodopeptides in urine help to discriminate between the various types of defects, and measurement of urinary iodide excretion also help to differentiate genetic defects from iodine deficiency or excess (Fig. 36.1 and Table 36.1). Since it is essential to treat newborn infants with CH immediately, blood and urine samples must be obtained immediately after referral. Radioiodide studies can be conducted a few days after T4 therapy is started, before the infant’s serum TSH concentration decreases to normal. Alternatively, these studies can be performed at 3 to 4 years of age, when T4 therapy can be interrupted safely for several weeks.
Defects in Ontogeny of the Thyroid Gland (Thyroid Dysgenesis)
Thyroid organogenesis (thyroidogenesis) consists of a series of partly sequential, partly synchronous developmental steps. Primordial thyrocytes that originate from a median placode in the ventral endoderm of the foregut (the floor of the future oral cavity) proliferate and migrate as a tubular structure to the definitive thyroid site in front of the second to fourth cartilage rings of the trachea. In the meantime, this median anlage diverges into two lateral lobes, each of which converge and fuse with the ultimobranchial bodies derived from the fourth and fifth branchial pouches. The anlagen are thought to fuse around the time that the organ reaches the definitive thyroid site in the neck. The primordial thyrocytes in the lobes further differentiate into thyroglobulin-producing cells and become arranged into follicles; primordial cells that originate from the neural crest and migrate in the lateral anlagen become the calcitonin-producing parafollicular cells (C-cells). Redundant tissue (i.e., most of the thyroglossal duct) is removed by apoptosis.
The embryonic phase of thyroidogenesis begins with the placode at about 3 weeks after fertilization, and ends with a bilobar solid organ at about 7 weeks of gestation. At approximately 12 weeks, the thyrocytes start arranging in follicles, secreting thyroglobulin and importing iodide into these developing spaces, and subsequently synthesize iodotyrosine and iodothyronine (mainly T4) residues in thyroglobulin. These early phases of development and maturation are TSH independent (see Chapter 2). After midgestation, the gradually growing thyroid progressively increases its secretion of T4, controlled by the fetal pituitary. Serum FT4 concentrations reach adult values at about 34 weeks of gestation (see Chapter 53).
Most patients with permanent CH-T, detected by neonatal screening, have some endogenous, presumably genetic, defect in thyroidogenesis (thyroid dysgenesis). The embryonic consequences, hampered migration and proliferation, but apparently not differentiation, can be visualized later in life (e.g., by radioiodide imaging) as thyroid rudiments with considerable variations in size, shape and anatomical site. The gland can be totally absent (agenesis), or, more often, stuck in an early phase of development (hypogenesis). The common feature of the spectrum of dysgenetic thyroid rudiments is that they are unable to fulfill the patients’ needs for T4, although the impressive T4-producing capacity of the rudimentary tissue demonstrates that its follicular architecture is intact, and the hormonogenesis is able to respond to elevated TSH levels. Thus it is the lack of tissue (hypoplasia) that is the limiting factor. Indeed, it is remarkable that even long-lasting exposure to high TSH levels does not generate catch-up growth (secondary hyperplasia), unlike the occurrence of goitrogenesis in untreated patients with thyroid dyshormonogenesis. In other words, in comparison with normal thyroid tissue, the responses of organogenesis and hormonogenesis to TSH stimulation are discordant in dysgenetic tissue.
Unfortunately, in the thyroid literature, the term “hypoplasia” is often used to describe a type of thyroid dysgenesis with the thyroid rudiment at the normal site in the neck. This might be due to misinterpretation of ultrasound images of the neck. Even in patients without any sign of T4-producing activity, and classified as thyroid agenesis, proven with the absence of radioiodide uptake and undetectable thyroglobulin levels in the circulation (see Table 36.1), some tissue may be present between the trachea and the carotid arteries. Yet, without any clue that this originates from primordial thyroid tissue, it should not be called thyroid hypoplasia. Also, a thyroid rudiment large enough to cover the full width of the trachea can be confused with a bilobar structure. We intend to reserve the term hypoplasia for a thyroid gland that underwent a normal embryonic and early fetal development, but lacked the normal TSH stimulation in the second semester of gestation, e.g., in case of CH-C, or the various types of TSH resistance. Whenever a thyroid gland has become stuck in its development, the rudiment is usually found along the virtual tract of the thyroglossal duct. For these sites, the term “dystopic” is more appropriate than “ectopic.”
The clinical consequences of CH-T due to thyroid dysgenesis depend entirely on the residual capacity of the rudiment to produce T4. Even tiny (usually sublingual) rudiments, strongly stimulated by TSH, appear to be able to secrete T4 in amounts that can conceal clinical manifestations for a long time. Larger dystopic rudiments may give raise to subclinical hypothyroidism (elevated serum TSH, and serum FT4 concentrations well within the reference range). The largest malformations, e.g., thyroid hemiagenesis, or supralingual rudiments are usually accompanied by TSH levels that remain below the cut-off levels used in neonatal screening programs.
While extensive and sophisticated animal studies have disclosed a number of transcription factors involved in thyroidogenesis (59), in only a small minority (less than 5%) of patients could associations between thyroid dysgenesis and mutated transcription factors (NKX2-1, NKX2-5, FOXE1, and PAX8) be established, and the percentage of familial cases is even
smaller (11,60). Indeed, thyroid dysgenesis is usually considered to be a sporadic disorder, but the higher prevalence among people of European origin than of African or Asian origin, and the higher prevalence among women (∼70%), suggest genetic involvement (61). Moreover, the fact that thyroid dysgenesis in monozygotic twins is usually discordant (11,62,63) is a major argument against any exogenous (maternal) factor as a primary cause. Presumably, combinations of defects in genetic conditions and exogenous factors involved in thyroidogenesis are responsible for the disturbed delicate balance between proliferation, differentiation, migration, bifurcation, fusion, and apoptosis that result in thyroid dysgenesis.
smaller (11,60). Indeed, thyroid dysgenesis is usually considered to be a sporadic disorder, but the higher prevalence among people of European origin than of African or Asian origin, and the higher prevalence among women (∼70%), suggest genetic involvement (61). Moreover, the fact that thyroid dysgenesis in monozygotic twins is usually discordant (11,62,63) is a major argument against any exogenous (maternal) factor as a primary cause. Presumably, combinations of defects in genetic conditions and exogenous factors involved in thyroidogenesis are responsible for the disturbed delicate balance between proliferation, differentiation, migration, bifurcation, fusion, and apoptosis that result in thyroid dysgenesis.
These processes cannot be considered apart from embryogenesis in general, although it is remarkable that thyroidogenesis can be severely disturbed without serious involvement of other organs or even adjacent structures. About 5% to 25% of the patients have additional congenital defects or phenotypic abnormalities, a significantly higher percentage than the control population [reviewed in (64,65)]. Reduction of the calcitonin-producing parafollicular cells (C-cells) is not included in these percentages.
The fate of the ultimobranchial bodies and C-cells when the median anlage is missing or is stuck in its development is poorly understood. In a recent case report, an adult patient was described with a sporadic medullary thyroid carcinoma in a lingual thyroid rudiment. Immunohistochemically, the tumor cells were positive for calcitonin. The patient was clinically euthyroid until the surgical removal of the tumor, and then she developed hypothyroidism. Indeed, no eutopic thyroid tissue appeared to be present (66). So, although the median thyroid anlage was stuck near its origin, at least some C-cells had migrated into the rudiment. On the other hand, adult CH patients with thyroid dysgenesis have decreased or undetectable serum calcitonin levels, and absent calcitonin-responses to intravenous calcium administration, in contrast to controls and CH patients with thyroid dyshormonogenesis (67,68). Serum calcitonin in infants with thyroid dysgenesis is decreased, and often becomes undetectable after the age of 3 (67), suggesting that maldevelopment of the thyroid is strictly associated with early onset of partial impairment of calcitonin secretion that can end in severe deficiency. Nevertheless, any negative influence on calcium homeostasis and bone mineral density in later life has not been observed (60,68).
Thyroid abnormalities recorded in thousands of autopsies have been studied to investigate the fate of the ultimobranchial contribution to human thyroid development (69). The prevalence of abnormalities is approximately 10 times higher than the prevalence of thyroid dysgenesis detected by neonatal screening. In half of the cases with a lingual thyroid, cystic structures up to 1.5 cm in diameter were noted in the region of the upper parathyroids. The cystic tissue stained positively for calcitonin.
The Brain–Thyroid–Lung syndrome is an illustrative example of syndromal occurrence of CH-T (70). It is clinically characterized by manifestations of brain dysfunction (including chorea, hypotonia, ataxia, and mental retardation; basal ganglia, cortical, and ventricular abnormalities), lung dysfunction (including infant respiratory distress and recurrent pulmonary infections), and CH-T, and it is genetically characterized by heterozygous NKX2-1 defects. Numerous published cases demonstrate the variability in the organs involved, in the severity, course and duration of the dysfunctions, and type of mutations (70,71,72,73,74). Many patients have de novo mutations, and in many of them the thyroid seems not to be affected, or the defect was too mild to be noticed. Whenever the thyroid is involved, it can be dysgenetic, varying from agenesis to hemiagenesis, or, more often, with a type of dyshormonogenesis that resembles TSH resistance, i.e., the thyroid’s morphology appears normal, without a tendency to goitrogenesis, with mild thyroid dysfunction (75). The syndrome has been fatal in a considerable percentage of patients with lung involvement (71).
The first patient diagnosed with the Brain–Thyroid–Lung syndrome had severe transient neonatal respiratory distress, mild permanent CH-T with a normally developed thyroid, neurologic problems and delayed mental and motor development. She had a de novo deletion of chromosome 14q13–21, which included the NKX2-1 gene (76). The next patients described, two sisters, had similar thyroid and brain dysfunction, but much more severe recurrent lung problems, being fatal in one of them. The siblings had a deletion of chromosome 14q12–13.3, and absence of the maternal NKX2-1 allele; their unaffected mother had a balanced translocation (77).
Many subsequent case reports have demonstrated that the syndrome is causally related to monoallelic inactivating mutations in NKX2-1 (73,78,79,80,81,82,83,84,85). In the thyroid anlage, NKX2-1 is involved in differentiation; in mature thyrocytes, NKX2-1 binds to the promoters of the various genes involved in TH production, up-regulating their transcription. Indeed, experiments in mice have shown that monoallelic inactivation of Nkx2-1 leads to half the level of TSHR and reduced thyroglobulin expression in the thyrocytes (86). The decreased TSHR activity is likely responsible for the elevated TSH concentration that indicates mild hypothyroidism, without compensation through goitrogenesis.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


